Суперпозиція ЛКАО і псевдопотенціалу для розрахунку енергетичної зонноїструктури монокристалів C
Суперпозиція ЛКАО і псевдопотенціалу для розрахунку енергетичної зонної
структури монокристалів CdJ2
Для розрахунку енергетичної зонної структури кристалів останнім часом набув поширення метод апріорних атомних псевдопотенціалів (ПП). У загальних рисах цей підхід ґрунтується на самоузгодженому пошуку ПП у наближенні функціонала локальної спінової густини. Стартова точка цієї процедури базується на релятивіському рівнянні Дірака для хвильової функції Gl(r) і Fl(r):
dFl(r)/dr - (g/r).Fl(r) + .[El - V(r)]Gl(r) = 0 (1a)
dGl(r)/dr + (g/r).Gl(r) - [(2/2) + El - V(r)].Fl(r) = 0, (1b)
де l = -1=137.07 – обернене значення константи надтонкої структури; g - ненульове ціле число. Розв’язки рівняння (1) визначають густину заряду:
(r) = [|Gl(r)|2 + | Fl(r)|2]. (2)
El < EF
Використовуючи процедуру нелінійної інтерполяції, визначимо псевдопотенціал:
Vps(l)(r) = Vост(r) + Vioн(l)(r)+ Vсо(l)(r), (3)
де Vост(r) – остовний потенціал, V(l)ioн(r) визначає іонну корекцію псевдопотенціалу і V(l)со(r) – спін-орбітальна корекція. Кожний з вищенаведених доданків може бути виражений в аналітичній формі і тому відповідні матричні елементи можна точно обчислити (замість числового інтегрування):
Vîńň(r) = (-Zv.e2/r).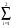 Ci.erf[i1/2.r]; (4)
Ci.erf[i1/2.r]; (4)
Vłîí(r) = 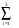 (Ai+r2.Ai+3).exp(-i.r2). (5)
(Ai+r2.Ai+3).exp(-i.r2). (5)
Інтерполяційні коефіціенти Ai, i, CI, i визначаються як розв`язки самоузгодженого рівняння Дірака – Хартрі – Фока – Слетера для конкретних атомів з відповідними орбітальними числами з подальшою нелінійною інтерполяцією (4, 5).
Повний псевдопотенціал є сумою нелокальних псевдопотенціалів, які перекриваються і розміщені в точках p,q. Взаємодія електрона з остовами, які описуються періодичними іонними ПП, визначається оператором:
Vps(r, r') = Vq,s(r - Rp - q,s, - Rp - q,s), (6)
p,q,s
де Rp – вектор прямої ґратки, який визначає розташування елементарної комірки; q,s – вектор, який визначає розташування s-го іона сорту q в елементарній комірці. При обчисленні матричних елементів секулярного рівняння в базисі плоских хвиль необхідно перейти від r-простору до оберненого G-простору за допомогою Фур’є перетворення:
<k+Gi|Vps(r, r’)|k+Gj>=N-1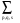 exp[-iq(Rp + q,s)].V-1.
exp[-iq(Rp + q,s)].V-1.
dr.dr’.exp[-i(k+Gj).r].Vq(r, r').exp[i(k+Gj).r'], (7)
де q = Gi - Gj і V – об’єм першої зони Бріллюена. Враховуючи, що qj.i = 2..ij (i, qj – основні вектори прямої й оберненої ґратки) знаходимо, що при довільних значеннях p exp(-iqRp) = 1, а тому сума по p просто дає множник N i остання рівність у локальному наближенні набуває вигляду:
<k+Gi|Vps(r, r’)|k+Gj>= exp[-iq.q,s)].V-1. d3r.exp[-i(q.r].Vq(r) (8)
q, s
Зауважимо, що тут інтегрування по r здійснюється в основній сфері кристала.
Розглянемо локальну частину псевдопотенціала. Форм-фактор потенціалу іона (4) дорівнює:
Vq = V-1. Vîńň(r).exp(-iqr).d3r.
Для обчислення цього виразу використаємо загальну процедуру. Запишемо розклад плоских хвиль за Релеєм:
exp(iqr) = 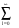 (2l+1).il.jl(|q.r|.Pl(cosq^r), (9)
(2l+1).il.jl(|q.r|.Pl(cosq^r), (9)
де jl(x) – сферичні функції Бесселя, Pl(cosq^r) – поліноми Лежандра l-го порядку і q^r – кут між q і r. Через сферичну симетрію s-орбіталі основний вклад в інтеграл у рівнянні (8) надає лише член ряду з l = 0. Враховуючи, що P0(cosq^r) = 1, одержимо:
2
V(q) = 4/V r2.{-Zve2/r) Ci.erf[i1/2.r]+(Ali+r2.Ali+3).exp(-li.r2)j0(qr)}d3r. (10)
0 i=1
Форм-фактор нелокальної частини псевдопотенціалу (перші два доданки 4) може бути визначений із:
< k + Gi|Vqíë (r, r’)| k + Gj > = V-1 d3r.d3r’.exp[-i(k + Gi).r].{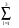 (Ail + r2.Ail+3).
(Ail + r2.Ail+3).
exp(-il.r2) - (A(0)l + r2.A(0)l+3).exp(-(0)l r2).Pl}.exp[-i(k + Gj).r]. (11)
Проекційний оператор l-ої компоненти моменту має вигляд:
Pl = 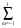 Ylm(i,).Y*lm(‘i,‘), (12)
Ylm(i,).Y*lm(‘i,‘), (12)
де Ylm(i,) – сферичні гармонічні. За визначенням, дія проекційного оператора на довільну функцію виражається так:
Pl exp(ikr) = Ylm(i,).{ d'.sin'.d'.Y*lm(‘,‘).exp(ikr)}, (13)
де , ', , ' – полярний і азимутальний кути векторів k i r. Використовуючи (13), рівняння (11) можна записати:
l 2
< k + Gi|Vqíë (r, r’)| k + Gj > = V-1 r2dr d sin d
m=-l 0 0 0
3
exp[-i(k + Gi).r].Ylm(,){ (Ail + r2.Ail+3).exp(-il.r2) - (A(0)l + r2.A(0)l+3).
l=1
2
exp(-0)l r2)}. d' sin'd Y*lm(‘,‘).exp[i(k + Gj).r] (11a)
0 0
Враховуючи умову ортонормованості сферичних функцій:
2 2
d sin.d. Ylm(, ).Y*l'm'(, ) = ll' .mm',
0 0
а також теорему додавання сферичних гармонік:
l
Ylm(, ).Y*lm(, ) = [-(2l+1)/4].Pl(cosGi^Gj),
m=-l
одержимо:
< k+Gi|Víë(r, r’)| k+Gj>= 4/V.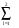 (2l+1).Pl(cosGi^Gj).Tl, (14)
(2l+1).Pl(cosGi^Gj).Tl, (14)
де
cosGi^Gj = GiGj/|Gi |.|Gj|, P0 = 1, P1 = cosGi^Gj, P2 = (3.cosGi^Gj - 1)/2;
а оператор Tl подається таким виразом:
Tl = 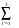 [All' M1l +All'+3.Mll' - A0l' M0l - A0l'+3.M0l']; (15)
[All' M1l +All'+3.Mll' - A0l' M0l - A0l'+3.M0l']; (15)
M1l = 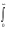 d3r.r2..jl(Gl.r).exp(-il r2). jl(Gj r); (16)
d3r.r2..jl(Gl.r).exp(-il r2). jl(Gj r); (16)
M2(l) = d3r.r4.jl(Gir) exp(-il r2).jl(Gj r). (17)
При проведенні самоузгоджених розрахунків враховувався екрануючий потенціал Хартрі-Слетера. Цей потенціал можна подати як звичайний кулонівський потенціал у формі, що визначається рівнянням Пуасона:
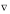 2.VH(r) = -4..e2.(r), (18)
2.VH(r) = -4..e2.(r), (18)
де (r) - електронна густина. Фур`є образ кулонівського потенціалу дорівнює :
VH(Gi - Gj) = [4..e2./|Gi - Gj|].(|Gi - Gj|) (19)
де
(Gi - Gj) = V-1 dr.(r).exp[-i(Gi - Gj).r] (20)
0
Крім заданого потенціалу іона Vlіон(r) та потенціалу Хартрі VH(r), необхідно також враховувати потенціал, пов`язаний з обміном та кореляцією електронів, які беруть участь в екрануванні. У літературі описані різні обмінно-кореляційні потенціали та наведено результати розрахунків, одержаних при їх застосуванні [1]. Найбільш поширеним є обмінно-кореляційний потенціал Слетера без урахування спінової поляризації електронних станів. Останні залежать від сорту атома і враховуються множником:
VOK(r) = -3e2..[(3/8.).(r)]1/3 (21)
Параметер змінюється в межах 0.5 - 1. Збільшення параметра посилює обмінно-кореляційний потенціал біля атома у порівнянні з міжатомною сферою. З цього випливає, що вибір обмінного потенціалу лише визначає розташування енергетичного спектра на шкалі енергій. Після перетворення Фур’є одержимо:
N
VOK(Gl - Gj) = -3e2..(3/8.)1/3.N-1 (rl)1/3.exp[-i(Gl - Gj).rl].
l = 1
Отже, матричні елементи ермітової матриці, яка визначає гамільтоніан, записуються наступним чином :
<k+Gi|H| k+Gj>=( k + Gi)2.Gi^Gj + 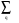 [v(q) (|Gi - Gj|) +
[v(q) (|Gi - Gj|) +
+< k + Gi|V(l)łîí| k+Gj>.(q).(Gi - Gj) + VH(Gi - Gj) + VOK(Gi - Gj) (22)
Структурний фактор іона типу q є:
(q)(|Gi - Gj|) = 1/2 exp[i.(|Gi - Gj|.p,q)].
p
Знаючи вираз гамільтоніана для старту процедури обчислення, необхідно вибрати певну початкову густину валентних електронів (r), яку подають як суперпозицію електронних густин нейтральних атомів:
(r) = Cq,n,l < n; q, l; n; q, l >,
n,q,l
де Cq,n,l – фактор заповнення орбіталі n, q, l атома n. Хвильова функція кожної орбіталі задається рівнянням:
q(r) = Rqnl(r).lm(, )/r.
Аналітичні вирази та коефіцієнти для функції Rqnl(r) наведені в роботах [2, 3].
Обчислення Фур’є перетворень для екрануючого кулонівського потенціалу VH(r) та обмінно-кореляційного потенціалу VOK[(r)], виходячи з функції Rqnl(r), дуже трудомісткі і, як показала практика, не завжди доцільні. Тому при проведенні обчислень замість суми локального потенціалу Vlq(q), кулонівського екрануючого потенціалу VH(q) та обмінно-кореляційного потенціалу VOK(q) використовувалася незалежна модель [4]:
Vq(q) = V(q)/(q),
де діелектрична проникність (q) визначається рівнянням:
(q) = 1 - [8..e2/Vŕň.q2].(1 - f(q)].(q).
Щоб отримати повну густину валентних електронів, треба додати (r) за всіма валентними зонами і за всіма різними станами k у першій зоні Бріллюена. Для визначення точного значення електронної густини здійснювалося додавання за багатьма точками ЗБ. Одержане значення використовується для обчислення Фур’є-образів кулонівського і обмінно-кореляційного потенціалів (19 - 21). Ітераційний процес самоузгодження зводився до розв’язування рівняння (7) на кожному етапі ітерації. Це дозволяло визначити густину валентних електронів, яка використовувалася на наступному етапі для обчислення екрануючого потенціалу. Показником ступеня самоузгодження є максимальна різниця власних значень гамільтоніана En,k на двох послідовних етапах ітерації. Точність розрахунків енергії була менша, ніж 0.2 eV, залежно від часу комп’ютерного розрахунку. Орбіталі йоду 5s-, 5px-, 5py-, 5pz і 5s-Cd вважалися базовими для секулярних рівнянь. Орбіталі 5s-J вводилися в першому порядку теорії збурень. Ефекти екранування приймалися до уваги для досягнення хорошого збігу між обчисленою енергетичною щілиною та її експериментальним значенням. Додаткове прискорення процедури самоузгодження досягалося внаслідок проведення варіації за параметром . Зміна параметра веде до зміни відповідного енергетичного терма і менше впливає на дисперсію.
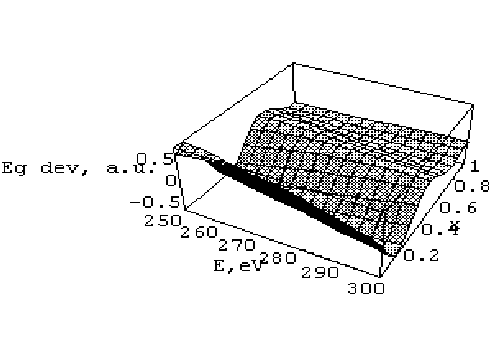
Рис 1. Залежність відносного відхилення Eg dev=(Egексп-Egобч.)/Egексп експериментальної ширини забороненої зони від обчисленої теоретично як функції енергії обрізування E
і параметра Слетера
Додаткова корекція (у випадку глибших зон) здійснювалася з використанням оптичних функцій, одержаних зі спектрів відбивання. Енергетична структура (включаючи третю координаційну сферу), розрахована в такий спосіб, в кінці була скоректована у відповідності до оптичних функцій для досягнення повного кількісного узгодження.
Проведені розрахунки показали, що метод ПП дає добрі результати для k-дисперсії зони провідності та для валентних станів. Більше того, з рис. 1 можна бачити, що обчислені значення ширини забороненої зони дуже чутливі до енергії обрізування і до параметра Слетера . Досягнуті методом псевдопотенціалу результати є суттєво нестабільними щодо енергії обрізування і обмінно-кореляційного параметера (див. рис. 1). З нашої точки зору, така нестабільність пов’язана з трудністю в описі заповнення валентних станів і структурою псевдопотенціалу.
Для спрощення і унаочнення розрахунків енергетичної зонної структури кристалів можна використати той факт, що практично у будь-якому структурному типі можна вибрати структурний фрагмент, який визначає основні параметри відповідних оптичних сприйнятливостей. На підставі кристалографічного аналізу й параметрів фононів структурні фрагменти (Cd2+J6-)-4 запропоновано вважати важливими для монокристалів CdJ2. У рамках такого підходу, згідно з виділеними структурними фрагментами, розв’язували задачу для цих фрагментів і лише потім враховували дію оточуючих кластерів. Для розв’язування секулярної задачі в рамках окремого структурного кластера найбільш доцільно застосовувати метод сильного зв’язку з використанням базису лінійних комбінацій атомних орбіталей (ЛКAO), центрованих на сусідніх атомах [5]. Для усунення будь-якого впливу граничних умов, здійснювалося перенормування зв’язків усередині кластера із застосуванням ефективної макроскопічної діелектричної сприйнятливості. Хвильова функція може бути виражена з використанням теореми Блоха для подання в реальному просторі:
k,n(r) = N-1/2 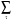 exp(ikn ).| n, r - n>, (22)
exp(ikn ).| n, r - n>, (22)
де |n> – атомні-орбіталі з симетрією n-типу ( тобто., s, p, d, ... ) і n – вектор, який визначає розташування n-го атома. Одноелектронна хвильова функція кристала звичайно записується у вигляді лінійної комбінації цих базових функцій:
| k > = 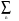 Cn | k, n>,
Cn | k, n>,
яку, звичайно, називають Блохівськими сумами. Границі додавання дуже важливі, оскільки вони визначають розмір кластера, а також вплив координаційних сфер при застосуванні теорії збурень. Метод ПП та похідні від нього з базисом плоских хвиль добре працює для слабко зв’язаних електронів, тоді як метод ЛКАО ефективний при сильній локалізації електронних станів. Це особливо стосується 3d-зон перехідних металів, а також більш глибоких станів валентної зони.
Усі матричні елементи гамільтоніана обчислювалися з використанням програмного пакета “Gaussian” для блохівських сум атомних орбіталей як базових функцій. Слід додати, що на цьому етапі для незайнятих станів було запропоновано скорегувати методом ЛКАО (процедура ортогоналізації) одержані методом ПП хвильові функції з метою досягнення найкращого узгодження з експериментальним значення енергетичної щілини. Тому в кожному випадку будувалася суперпозиція стартових базових орбіталей, одержаних як суперпозиція ПП і ЛКАО методу з відповідним ваговим множником. У такий спосіб вдалося використати переваги обох методів.
Остовні стани не включалися в базис, щоб обмежити максимальний розмір секулярного рівняння. Нехтування остовними станами в методі ЛКАО може створювати серйозні ускладнення при обчисленні власних значень енергії, оскільки власні значення енергії мають тенденцію сходитися до остовних станів замість валентних.
Ортогоналізація валентних орбіталей до остовних станів дозволяла обмежити розмір гамільтоніанової матриці до 568. Задана базова розмірність потім збільшувалася майже до 1960 з енергією обрізування до 106 Рідберга. Головний ітераційний критерій досягнення стабілізації власних станів полягав у збігу двох сусідніх власних значень енергії з точністю 0.02 Рідберга.
Дійсні орбіталі вільних атомів замінювалися певними локальними функціями слетерівського типу, котрі дуже подібні до дійсних іонних хвильових функцій, обчислених за першими принципами, хоча перші визначають дисторсію електронного вкладу. Підстановка одержаних у такий спосіб оптимізованих орбіталей дозволяла проводити числову оцінку з використанням техніки скорочення (стиснення) Гаусіана. Ми збудували ортогоналізовані Блохівські суми в такому вигляді:
Bq’a(k,r) = Bqa(k,r) + 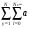 qa,lgBlg (k,r), (23)
qa,lgBlg (k,r), (23)
де Blg(k,r) хвильові функції зон провідності, одержані методом ПП. Всі aqa,lg і Blg, обчислювалися відповідно до умов ортогональності попередньо обчислених нормованих хвильових функцій ПП.
Потенціал одноелектронного гамільтоніана виражався у вигляді суперпозиції атомних потенціалів Va(r). Атомний потенціал апроксимувався наступним рівнянням:
Va(r) = (- Zve/r) 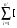 ci.exp(-air2) + Air2.exp(-bir2)] , (24)
ci.exp(-air2) + Air2.exp(-bir2)] , (24)
де всі пошукові коефіціенти ci, ai, Ai і bi обчислювалися за допомогою нелінійної інтерполяційної процедури. Використовуючи від восьми до дванадцяти гаусіанів, вдавалося забезпечити добрий хід радіальних функцій у наших обчисленнях. Усі матричні елементи гамільноніана розбивалися на серії з трицентрових інтегралів, котрі включали два гаусіани, центровані в точці розміщення атомів A і B, та атомний потенціал навколо точки C. Додавання здійснювалися шляхом розв’язування рівняння:
{ Hij(k) - E(k)Sij } = 0, (25)
для різних точок ЗБ. Матричні елементи слід обчислювати з більшою точністю, ніж це необхідно для обчислення власних значень через велику розмірність одержаного секулярного рівняння. Сумування велося за дванадцятьма сусідніми вузлами. Числове інтегрування здійснювалося в реальному просторі з урахуванням вкладу електрон-електронної взаємодії. Приклади інтегралів перекриття для s- і p-станів визначалося наступним рівнянням:
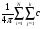 icj[p/(ai + bj)]3/2exp{[-aibj/(ai + bj)](B - A)2} (26)
icj[p/(ai + bj)]3/2exp{[-aibj/(ai + bj)](B - A)2} (26)
sa|pxb> =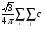 i cj {[p/(ai+bj)](B-A)2}exp{[-aibj/(ai+bj)](B-A)2}DBx (27)
i cj {[p/(ai+bj)](B-A)2}exp{[-aibj/(ai+bj)](B-A)2}DBx (27)
Матричні елементи операторів Хартрі-Фока мають вигляд:
Fij = cicj [p/(ai + bj)]3/2exp{[-aibj/(ai + bj)](B - A)2 }. (28)
Точка D, що визначає розміщення центра мас атомів A і B, визначається так:
D = (aiA + bjB)/(ai + bj) , (29)
Ефект екранування враховувався через модельні поправки Пердю-Зунгера і Капелі-Альдера [6] в такому вигляді:
mxc = - 0.6193/rS - 0.14392/(1+1.0529rS1/2 +0.3334rS).{1 +
+ [(0.5264rS1/2 +0.3334rS)/(3.(1+1.0529rS1/2 +0.3334rS))]}, (30a)
для rS > 1
mxc = - 0.6193/rS +0.031 ln(rS) - 0.0583, (30b)
для rS < 1; де rS = [3/(4)]1/3 ; – електронна густина.
Самоузгодження досягалося прямою ітераційною процедурою. Для скорочення числа ітерацій і забеспечення збіжності застосовувалося змішуванням електронної густини (m-1)-ї ітерації з 60 % початкової перед їх підстановкою в наступне рівняння. Відповідний екрануючий потенціал будувався зі застосуванням наближення Томаса-Фермі, що дозволяло позбутися окремих помилок при обчисленні електронної густини. Критерій самоузгодження зарядової густини вимагає:
| âčő,m - âő,m | < (31)
після m-го ітераційного кроку. Досягнута точність, менша ніж = 0,07% між вхідними і вихідними параметрами ітераційного кроку, служила головним критерієм самоузгодження. Власні значення енергії були стабільними потужністю до 0,003 атомних одиниць енергії. Процедура діагоналізації гамільтоніана здійснювалася QL методом [7].
Для підвищення точності опису густини електронних станів діагоналізація гамільтоніану здійснювалася для 64 рівновіддалених точок в 1/16-ій частині ЗБ. Числові обчислення проводилися з використанням методу теадраедрів. Вираз для інтегралів перекриття, які використовувалися при обчисленні:
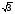 /4
/4 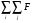 ijDBy
ijDBy (32)
(32)
/4 ijDBz (33)
/4 ijDAx (34)
/4 ijDAxy (35)
/4 ijDAz [DAxDBx + 1/2(ai + bj )] (36)
ijDAz [DAxDBx + 1/2(ai + bj )] (37)
ijDAxDBy (38)
ijDAxDBz (39)
ijDAyDBx (40)
ij [DAyDBy + 1/2(ai + bj )] (41)
ijDAyDBz (42)
ijDAzDBx (43)
ijDAzDBy (44)
ij [DAzDBz + 1/2(ai + bj )], (45)
де DBx = Dx - Bx ł DAx = Dx - Ax.
Після подібної процедури як і у випадку плоских хвиль, можна отримати кінцеві вирази для матричних елементів секулярного рівняння:
<k,q| H - E(k) | k,q’>= [ Ea - E(k) ] [ Sqq'’+ 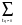 exp(-ikq ).Sqq’ +
exp(-ikq ).Sqq’ +
Bqq' + 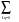 exp(-ikq )Aqq’, (47)
exp(-ikq )Aqq’, (47)
де
Sqq’ = < n | n’, r + l - l’ > ; (48a)
Bqq’ = < n | V(r ) - Va(r) | n’ > ; (48b)
Aqq’ = < n | V(r ) - Va(r) | n’, r + l - l’ > (48c)
Зауважимо, що всі складності цього методу зводяться до обчислення інтегралів (48).
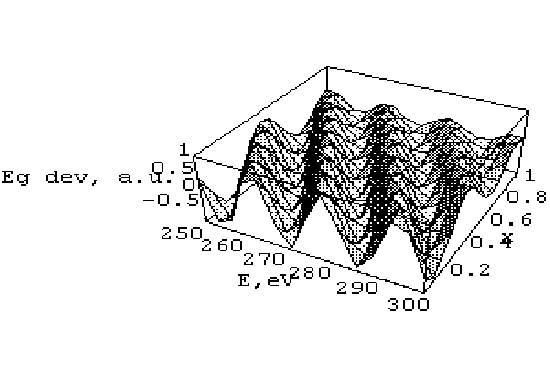
На рис. 2 наведено відхилення енергетичної щілини від енергії обрізування і ефективного параметра . Стабільність одержаних результатів суттєво вища, ніж для методу ПП. Тому останній базис було використано для обчислення електронної зонної структури відповідних твердих розчинів.
Рис 2. Залежність відносного відхилення Eg як функція параметра Слетера
та енергії обрізування. Позначення ті ж, що і на рис. 1
Із рис. 2 видно зростання стабільності одержаних результатів. Максимальна відповідність (до 1.5 eV ) є необхідною для встановлення належності електронного зв’язку до октаедра CdJ6. Дисперсія зонної структури досліджувалася для вершини валентної зони вздовж напрямку G-X-S, що визначається p-станами аніона J. Валентна зона, утворена pJ-орбіталями, розміщена нижче. Стани sJ є основними для майже бездисперсійної зони, яка лежить на відстані 3eV нижче pJ зони. Дно зони провідності (майже бездисперсійне) утворене антизв’язуючими 5s-орбіталями Cd. Антизв’язуючі p-орбіталі J i 3d-орбіталі Me беруть участь у формуванні наступної зони (з енергіями вище за попередню на 4 eV). Кластери CdJ6 формують вищу енергетичну зону, яка зазвичай не бере участі в оптичних переходах.
На основі одержаних хвильових функцій проведено розрахунки електронної густини як суперпозиції відповідних густин на окремих атомах:
(r) = *(r, k).*(r, k)

Рис 3. Розподіл електронної густини поблизу Cu центрів у монокристалах CdJ2
На рис. 3 наведено вклад нецентросиметричного розподілу електронної густини поблизу центрів Cu для монокристалів CdJ2-Cu. Із рисунка видно, що асиметрія розподілу заряду спричиняє специфічність міжшарової взаємодії між конкретним локальним центром і асиметрією шаруватої матриці кристала. Ця асиметрія відображає можливість передачі заряду між локальними і нелокальними центрами. Можна стверджувати, що тип нецентросиметричності дуже важливий для пояснення нелінійно-оптичних властивостей згаданих центрів. Дуже важливо, що кристалічна система дуже чутлива до локального розупорядкування мідних центрів. Структурні фрагменти CdJ6 і CuJ4 в монокристалах CdJ2-Cu відповідають в основному ковалентним зв’язкам, хоча фрагменти CdJ6 і CuJ4 у своїй основі є іонними. Ковалентний зв’язок визначається сильною pJ-pJ гібридизацією внутрішарових орбіталей з низькою симетрією в напрямку до атомів Cu (див. рис. 3). Орбіталі 5pJ визначають зв’язок між різними кластерами системи CdJ2-Cu як такої.
Гібридизація між структурними компонентами CdJ6 і CuJ4 суттєво зменшується, і тому поляризаційність хімічних зв’язків зростає внаслідок перерозподілу електронної густини між 5s-Cd і 5p-J орбіталями. Вклад у зв’язки 5s-J станів можна знехтувати.
Література
Бассани Ф., Дж. Пастори Паравичини Электронные состояния и оптические переходы в твердых телах.– М.: Наука, 1982.
Clementi E., Roetti C. Roothaan Hartree-Fock Atomic Wave Functions // Atomic data and nuclear tables.– 1974.– V.14.– P. 177-478.
Mclean A.D. Roothaan Hartree-Fock Atomic Wave Functions. Slater basis-set expansions for Z=55-92. // Atomic data and nuclear tables.– 1981.– V.26.– P.197-381.
Incson J. To the exchange-correlation theory. Sydney.– 1987.
Довгий Я.О., Китык И.В., Маньковская И.Г. Рентгеновские эммиссионные спектры монокрис-таллов прустита // ФТТ.– 1990.– Т.32.– №10.– С. 3170-3171.
Лобач В.В. К теории расчета зон ионных кристаллов.– Свердловск, 1989.
Щуп Т.Е. Прикладные численные методы в физике и технике.– М.: Высш. шк.– 1990.

Нравится материал? Поддержи автора!
Ещё документы из категории астрономия :
Чтобы скачать документ, порекомендуйте, пожалуйста, его своим друзьям в любой соц. сети.
После чего кнопка «СКАЧАТЬ» станет доступной!
Кнопочки находятся чуть ниже. Спасибо!
Кнопки:
Скачать документ