Плазменные технологии
В.И. БОРОДИН
ПЛАЗМенные ТЕХНОЛОГИИ
1. Плазменные технологии (области применения, преимущества, недостатки, перспективы)
2. Термодинамика и кинетика плазмохимических процессов
2.1 Термодинамический анализ плазмохимических систем
2.2 Кинетика плазмохимических процессов
2.2.1 Кинетика равновесных процессов
2.2.2 Неравновесные плазмохимические процессы
3. Технологическое оформление плазмохимических процессов
3.1 Плазмохимический реактор
3.1.1 Функции распределения по временам пребывания
3.1.2 Характеристики реального реактора вытеснения
3.2 Перемешивание реагентов с энергоносителем в смесителе
3.2.1 Стадии перемешивания
3.2.2 Модель перемешивания газовых потоков в цилиндрических каналах
3.3 Закалка продуктов плазмохимических процессов. Способы закалки
Список литературы
1. Плазменные технологии (области применения, преимущества, недостатки, перспективы)
Одной из основных тенденций развития современной техники и технологии является все более широкое использование высоких параметров технологических процессов: температуры, скорости, времени контакта и т.д.
Обусловлено это, в основном, следующими факторами развития цивилизации в последние десятилетия.
С одной стороны, основные производства, обеспечивающие человечество традиционными многотоннажными продуктами (энергоносителями, металлом, пластмассами и др.), сформировались, в основном, на базе научных концепций XIX – начала XX века или еще более раннего времени (например, металлургия) и обладают рядом недостатков, связанных с многостадийностью, громоздкостью, расточительностью по энерго- и ресурсозатратам, а также большими отходами. Большинство технологий в рамках традиционных подходов достигли своего критического состояния.
Дальнейшее развитие промышленной базы с использованием таких подходов невозможно, так как влечет за собой необоснованное наращивание объема отдельных производств, неоправданные затраты ресурсов для создания производственных площадок и оборудования, быстрое истощение полезных ископаемых, существенное ухудшение экологической обстановки.
С другой стороны, во II половине XX века началось бурное развитие новых отраслей (атомной и реактивной техники, электроники и др.), что потребовало значительного качественного улучшения традиционно используемых материалов, а также создания новых материалов, уникальных по своим свойствам (чистоте, термической и химической стойкости, твердости и т.д.).
Все это предопределило интенсивную работу по поиску и разработке новых технологических решений в металлургии, химической промышленности, энергетике, машиностроении и др.
Одним из путей решения данных проблем является существенное повышение температуры, при которой реализуется тот или иной технологический процесс. Указанные причины привели к возникновению и развитию нового направления физической химии и химической технологии плазмохимии (ПХ), где осуществление химических реакций происходит в высокотемпературной среде плазмы или созданной плазмой.
Для получения низкотемпературной плазмы (с точки зрения химии плазма является высокотемпературной, так как имеет температуру порядка 103 ÷ 105) используются различные способы. Наиболее простым и широко применяемым способом является электрический разряд в газе – так называемая газоразрядная плазма.
При прохождении электрического тока через газовую среду энергия электрического поля, созданного внешним источником тока, преобразуется во внутреннюю энергию газа. За счет этого, во-первых, повышается его температура, во-вторых, происходит распад атомов и молекул газа на радикалы, ионы, электроны. Происходит возбуждение частиц плазмы, которые затем излучают кванты различного излучения в диапазоне от инфракрасного до ультрафиолетового.
В результате в среде, находящейся в состоянии плазмы, за счет повышения температуры Т значительно повышаются скорости химических реакций w, поскольку, по формуле Аррениуса, w ~ exp(-Ea/T).
Кроме того, в плазме, из-за появления высокореакционных частиц и излучения, возникают физико-химические взаимодействия, которые могут приводить к образованию новых соединений, не образующихся в обычном (низкотемпературном) состоянии (например, соединений из инертных газов). Более того, традиционные химические продукты, полученные в плазме, часто отличаются по своим свойствам от тех же продуктов, но полученных при обычных температурах.
Таким образом, перевод веществ в состояние плазмы увеличивает их реакционную способность.
В зависимости от свойств плазмообразующей среды и условий реализации разряда, состояние плазмы может иметь существенное различие. В частности, состояние плазмы может быть достаточно близким к состоянию термодинамического равновесия (квазиравновесие), а может быть далеким от него (неравновесная плазма). Поэтому ПХ-системы иногда условно разделяют на два больших класса – неравновесные и квазиравновесные.
Состояние квазиравновесной плазмы, характеризующееся одной температурой, можно достаточно точно описывать с помощью равновесной термодинамики (ТД) и статистической физики. Это позволяет предсказать и рассчитать различные параметры, а также продукты химических процессов, которые образуются в такой плазме, не вдаваясь в суть происходящих элементарных явлений, которые в данном случае описываются классической равновесной химической кинетикой.
Гораздо более богатой по реакционным возможностям является неравновесная плазма, которую нельзя характеризовать одной температурой. В такой плазме каждый сорт частиц и их состояние можно характеризовать своей температурой, например, температура электронов, температура ионов, вращательная температура, колебательная температура, температура заселения и др. При этом надо отметить, что здесь «температура» является просто удобным параметром, позволяющим использовать при расчетах известные статистические функции распределения (функции Максвелла, Больцмана, Саха, Планка и др.) для соответствующего рода частиц. Эти температуры могут сильно отличаться друг от друга. Например, в разряде низкого давления температура электронов (десятки тысяч градусов) может быть в сотни раз больше температуры атомов и ионов (сотни градусов).
В неравновесной плазме можно получить самые экзотические продукты (например, различные соединения инертных газов).
Как правило, неравновесная плазма получается при низких давлениях плазмообразующей среды (Р ≤ 100 мм рт. ст.), а квазиравновесная – при высоких давлениях (P ≥ 1 атм).
Для получения плазмы используют все виды электромагнитных полей: постоянное, переменное, высокочастотное (ВЧ), сверхвысокочастотное (СВЧ), оптическое излучение и т.д., а также различных типов разрядов: дуговой, тлеющий, коронный и др. Свойства плазмы, способы и устройства для ее получения (генераторы плазмы) подробно описаны в литературе [1–7].
Основные диапазоны изменения параметров плазмы, используемой в технологиях, составляют по давлению от 10-5 мм рт. ст. до 200 атм, по температуре T = 103 ÷ 105 K.
Рассмотрим примеры конкретных технологических применений плазмы.
Неравновесные ПХ-процессы, в основном, используются для обработки поверхностей различных материалов: модификация поверхностей, нанесение и снятие пленок и т.д. Все эти процессы являются малотоннажными. Процессы проводят, как правило, в тлеющем разряде при давлениях плазмообразующей среды порядка или меньших 1÷10 мм рт. ст.
Плазмохимическая модификация поверхностей. Под модификацией поверхности твердого тела подразумевают изменение химического состава и структуры поверхностного слоя.
ПХ-обработка позволяет получить материалы с уникальными свойствами поверхности, обусловленными одновременным воздействием излучения плазмы и химически активных частиц при относительно низкой температуре поверхности.
Блок
пита-
ния
Откачка
Сис-тема
газо-снабже-ния
Де-таль
Тлеющий разряд
+
–
Рис. 1. Схема установки для модифицирования поверхности металлов в тлеющем разряде
ПХ-обработке могут подвергаться различные материалы: от металлов и их сплавов до различных полимеров как естественного, так и искусственного происхождения.
ПХ-обработка металлов и сплавов. Типичная схема установки для модифицирования поверхностей металлов в плазме тлеющего разряда приведена на рис. 1 [8]. В откачиваемую металлическую камеру помещают обрабатываемую деталь. В пространстве между поверхностями детали и стенки зажигается тлеющий разряд. Состав и давление плазмообразующего газа легко регулируется системой газоснабжения.
Образующиеся в плазме заряженные частицы бомбардируют поверхность детали, в результате чего может изменяться химическая и кристаллическая структура поверхности и ее свойства (твердость, химическая и термическая стойкость и др.).
Например, в машиностроении широко используется процесс азотирования поверхности деталей, когда в качестве плазмообразующего газа используется азот. Бомбардировка поверхности детали происходит ионами азота, поверхность насыщается азотом, образуя нитрид железа. В результате на поверхности детали образуется слой, обладающий высокой твердостью и коррозионной стойкостью.
Азотированию подвергаются, в основном, трущиеся детали (валы, штоки и др.), что позволяет увеличить ресурс их работы в несколько раз. Наиболее крупные установки для азотирования (США, Франция) позволяют обрабатывать валы диаметром до 2 м и длиной до 12 м. Мощность таких установок достигает 300 кВт при давлении Р = 0.1–10 торр и напряжении U = 500 –1500 B. Время обработки варьируется от 10 минут до 20 часов, при этом толщина азотированного слоя изменяется от 30 мкм до 0.7 мм.
ПХ-обработка органических материалов. При обработке плазмой органических материалов как искусственного, так и естественного происхождения (полимеров, пластмасс, тканей, бумаги и др.) изменяется состав и реакционная способность их поверхностей, что приводит к изменению их свойств.
Так, при обработке тканей может изменяться адгезия, смачиваемость, окрашиваемость, возможность склеивания как между собой, так и с другими материалами. Может резко уменьшиться усадка обработанных волокон шерсти, измениться их электростатические характеристики и др.
Обработка органических материалов может производиться в вакуумированных объемах по типу установки, приведенной на рис. 1, а также при высоких давлениях.
Полотно
Диэлектрик
Элект-род
ВЧ-генератор
Зазор
Рис. 2. Схема устройства для обработки рулонного полотна коронным разрядом
Вал
На рис. 2 [9] приведена схема установки для обработки рулонного материала в плазме коронного разряда. Плазма горит в зазоре под внешним электродом, воздействуя на движущееся полотно. Обработка натуральных полимеров в плазме коронного разряда приводит к увеличению сцепления волокон между собой, а также с другими материалами. Так, после обработки в кислородной плазме сила сцепления пластин из целлюлозы друг с другом, а также с полимерными пленками возрастает в 5–7 раз.
Получение и снятие пленок и покрытий. С помощью плазмы можно получать и снимать как неорганические, так и органические пленки, что широко используется в различных отраслях промышленности, науки и техники.
Так, например, плазменные методы осаждения и травления пленок широко используются в микроэлектронике при изготовлении элементов тонкопленочных интегральных схем.
На рис. 3 [10] приведена схема плазменной обработки кремниевых пластин в микроэлектронике.
Через объем ПХ-реактора с обрабатываемыми пластинами пропускается плазмообразующий газ, соответствующий заданной технологической операции. Плазма при давлениях порядка 0.1–10 мм рт. ст. создается с помощью индуктора (катушки), надетого на реактор и питаемого от ВЧ-генератора.
В данной схеме можно проводить различные технологические операции с кремниевыми пластинами: плазменная очистка поверхностей, ПХ-травление и удаление фоторезиста, осаждение диэлектрических пленок и антикоррозионных (пассивирующих) покрытий.
Переход от одной операции к другой производится заменой состава продуваемого плазмообразующего газа. В качестве рабочих газов при химической очистке и травлении чаще всего используют различные органические галогенсодержащие соединения, например, CHCl3, CHCl2F, CHClF2 и др. [11].
Плазменные («сухие») методы обработки при производстве элементов электронной техники используют сравнительно дешевые газообразные реагенты, имеют высокую производительность и легко поддаются автоматизации.
Рис. 3. Схема плазмохимического удаления фоторезиста
В кратком пособии невозможно описать все разнообразие пленок и покрытий, получаемых с помощью плазмы. Отметим только два из них – покрытия нитридом титана и тефлоном. Нитрид титана (TiN) является очень твердым, термо- и химически стойким веществом и широко используется для покрытия режущих элементов различных инструментов (в том числе сверл, фрез, буров и т.д.), а также различных поверхностей, работающих в химически агрессивных средах (в том числе искусственных зубов в стоматологии). Обладая красивым золотистым цветом, TiN используется для декоративной отделки помещений («золотые» кафельные плитки).
В различных технических устройствах широко используются резинотехнические изделия (манжеты, сальники и т.д.), которые должны легко скользить по сопряженным плоскостям. Сама резина этим свойством не обладает, поэтому покрытие таких изделий материалом с малым значением коэффициента трения – тефлоном, приводит к существенному увеличению износостойкости изделия, тем более что тефлон предохраняет их от воздействия агрессивных сред.
Плазмой обрабатываются не только твердые материалы, но и различные жидкости. При этом, в отличие от твердых тел, в жидкости могут возникать значительные потоки переноса массы (диффузия, конвекция), в результате чего возникшие при взаимодействии с плазмой физико-химические изменения в поверхностном слое жидкости могут распространиться по всему ее объему. Таким образом, за счет процессов перемешивания обработку объема жидкости можно проводить через обработку ее поверхности.
В качестве примера такого процесса можно отметить обработку плазмой воды с целью ее очистки и обеззараживания [12]. Обеззараживание воды плазменной обработкой может служить альтернативой процессу хлорирования, поскольку при хлорировании воды образуются хлор-органические соединения, обладающие канцерогенными свойствами.
Плазмохимические методы переработки материалов. Под ПХ-переработкой подразумеваются процессы, в которых все исходное вещество (а не только его поверхность) проходит стадию высокотемпературного состояния, обусловленного взаимодействием с плазмой, а химические реакции осуществляются, в основном, в газовой фазе. Из-за необходимости иметь высокую производительность в многотоннажных технологиях используется, как правило, квазиравновесная плазма высокого давления, которая получается в специальных устройствах – генераторах плазмы (плазмотронах).
Схема организации ПХ-переработки материалов приведена на рис. 4. Перед тем как попасть в ПХ-реактор, исходное сырье претерпе-вает некоторую обработку, среди которой следует отметить измельчение (диспергацию). Чем более диспергировано вещество, тем более эффективна переработка, поскольку нагрев и испарение более мелких частиц происходит быстрее и с меньшими затратами энергии.
Из узла подготовки сырье поступает в ПХ-реактор, где перемешивается с плазмой, поступающей из плазмотрона, нагревается, испаряется, вступает в различного рода химические реакции. Нередко плазма образуется в том же ПХ-реакторе (совмещенный реактор-плазмотрон).
Подготовка сырья
ПХ-реактор
Закалка
Разделение смеси
Плазмотрон
Рис. 4. Схема технологии плазмохимической переработки сырья
Продукты реакций охлаждаются и разделяются. Часто охлаждение производится с очень высокой скоростью (закалка): 104–108 К/с . Задача закалки состоит в сохранении тех продуктов, которые получились при высоких температурах и которые при охлаждении могут либо разложиться, либо вступить в другие химические реакции в процессе охлаждения.
С помощью закалки можно получать вещества в метастабильном состоянии, отличающемся от нормального состояния (структурой, электрическими, термическими и другими свойствами).
ПХ-переработка органического сырья и отходов.
В литературе описаны результаты сотен исследований по взаимодействию плазмы с различными органическими веществами разной глубины и различной степени проработанности для целей организации производства. Однако даже основная масса проработанных и подготовленных для многотоннажной производственной реализации процессов до сих пор не реализованы. И это, в первую очередь, обусловлено основным недостатком ПХ-технологий – большим потреблением электрической энергии, которое делает их в ряде случаев пока не конкурентоспособ-ными с другими технологиями.
И, тем не менее, в мире существуют, а также находятся в стадии разработки большое количество технологий ПХ-переработки органических материалов для различных целей: получения мономеров для синтеза (включая часто и сам синтез) полимеров, пластмасс, энергоносителей, восстановителей и др. При этом в качестве исходного продукта используются не только добываемое сырье земных недр, но и возобновляемое растительное сырье, а также промышленные и бытовые отходы.
Все многотоннажные процессы ПХ-переработки органических материалов можно условно разделить на традиционные (классические) и нетрадиционные. К традиционным можно отнести процессы, давно разработанные и промышленно реализованные еще в первой половине ХХ столетия, которые, однако, постоянно совершенствуются исходя из конкретной ситуации, складывающейся в мире в тот или иной промежуток времени.
Первым направлением традиционной многотоннажной ПХ-технологии является производство ацетилена, который является сырьем для производства разнообразных продуктов основного органического синтеза, а также используется в некоторых других отраслях промыш-ленности.
ПХ-производство ацетилена.
Бурное развитие производства ацетилена и ПХ-метода его получения произошло в 40-е и 50-е годы ХХ столетия. Многотоннажный органический синтез в мире (особенно в Европе) до 60-х годов в основном базировался на ацетилене. Были построены десятки заводов по его производству плазмохимическим способом.
Однако в 60-е годы были разработаны способы получения тех же продуктов из относительно дешевого этилена. В результате основная масса заводов по производству ацетилена была закрыта, а его роль в 70-е годы стала незначительной.
Но с течением времени ситуация в мире постепенно меняется в пользу ацетилена, поскольку этилен вместе с его источником производства – нефтью быстро дорожает. Интерес к ацетилену как исходному сырью для органического синтеза постоянно возрастает и считается, что через несколько десятилетий ацетилен может занять снова доминирующее положение в органическом синтезе.
Следует отметить, что для производства ацетилена сырьевая база гораздо шире, чем для этилена, и его можно эффективно получать не только из нефти или газа, но и из угля, а также из растительного сырья и органических промышленных и бытовых отходов.
В промышленных масштабах ПХ-способ получения ацетилена осуществляют пиролизом углеводородов, либо непосредственно в дуговом разряде (электрокрекинг), либо в плазменной струе водорода, нагреваемой в плазмотроне. Получение ацетилена в промышленных масштабах впервые было осуществлено в Германии на заводе в г. Хюльсе в 1940 году с производительностью 60 тыс. т ацетилена в год, который работает до сих пор.
В связи с постоянным удорожанием используемого для производства ацетилена сырья – нефти и газа, а также с перспективой их исчерпания в обозримом будущем в мире достаточно интенсивно разрабатываются ПХ-технологии, базирующиеся на более распространенном и менее дефицитном сырье – угле и углесодержащих промышленных отходах.
Вторым классическим (традиционным) направлением ПХ-переработки органических материалов является конверсия их в синтез-газ (конвертированный газ), состоящий из СО и Н2.
Синтез-газ широко используется в различных химических процессах при производстве аммиака, метанола, высших спиртов и других химических продуктов, а также жидкого топлива, заменяющего бензин и дизельное топливо. Его применяют как восстановитель при получении металлов и других соединений из оксидного, сульфидного и другого сырья.
При добавлении к углеводородам (система С–Н) кислородсодержащих соединений происходит интенсивное образование соединения СО, которое становится превалирующим в системе С–Н–О, поскольку из всех молекул молекула СО имеет наибольшую энергию связи.
В отличие от ацетилена, синтез-газ (СО + Н2) термодинамически устойчив и при комнатной температуре, поэтому получить его технологически гораздо проще и дешевле – отпадает необходимость в закалке пиролизного газа и поддержании слишком высоких температур процесса.
Углеродсодержащим сырьем для получения синтез-газа может служить любой вид материалов, содержащий углерод: газообразные и жидкие углеводороды, уголь, торф, биомасса, промышленные и бытовые отходы. Для стехиометричности состава в качестве недостающего кислородсодержащего сырья используются вода (водяной пар), двуокись углерода, воздух и кислород.
В принципе синтез-газ получают без всякой плазмы обычным процессом сжигания. Однако в отличие от таких методов плазменный метод позволяет обеспечить низкое содержание СО2, Н2О в продуктах конверсии или полное их отсутствие и таким образом исключить дорогостоящую операцию очистки там, где синтез-газ требуется достаточно чистым, например, в органическом синтезе, в металлургии и др.
При плазменном пиролизе, благодаря высоким температурам, реализуемым в зоне реактора, продукты пиролиза состоят из газовой составляющей. Степень превращения сырья в целевые продукты при этом близка к 100%. Причем в газообразных продуктах отсутствуют смолы, фенолы и углеводороды, загрязняющие продукты обычной газификации сырья.
Для конверсии органического сырья в синтез-газ используются технологические схемы, подобные приведенной на рис. 4, но без узла закалки. Поскольку время нагрева сырья здесь не лимитируется, то нет необходимости создавать высокие скорости прогрева частиц сырья, что является серьезной проблемой при получении ацетилена, да, зачастую, и нет необходимости до такой степени измельчать исходное сырье.
Поскольку температуры конверсии не очень велики, то для реализации процесса в качестве ПХ-реакторов часто используют достаточно вместительные емкости, обычно футерованные внутри высокотемпературными материалами, выдерживающими рабочие температуры процесса (рис. 5 [6] и 6 [13]).
Рис. 5. ПХ-реактор с расплавом стали: 1–4 – подача угля (размер зерен 1.8 см), воды, азота и воды соответственно; 5 – расплав стали; 6 – зона выделения летучих; 7 – пар; 8 – газовый продукт
8
Рис. 6. Плазменный газификатор шахтного типа
Плазменный газификатор, схема которого приведена на рис. 6, используется для получения газа-восстановителя при производстве губчатого железа в процессе Plasmared, разработанном шведской фирмой SKF Steel Engineering AB. В первом варианте для получения восстановительного газа использовали только уголь и водяной пар (реакция С + Н2О → →СО + Н2).
В дальнейшем фирма SKF разработала процесс получения конвертированного газа высокой калорийности из различных углесодержащих соединений: торфа, биомассы, отработанных масел, угля, бытовых и промышленных отходов.
Фирма «Плазма энерджи» (США) также разработала ПХ-процесс получения топливного газа из угля, торфа и различных органических отходов (в том числе из старых автопокрышек). Стоимость такого топлива сопоставима со стоимостью импортируемой в США нефти.
Наиболее крупное производство конвертированного газа из угля и биомассы для восстановления губчатого железа реализовано в ЮАР немецкой фирмой «Хюльс» (более 250 тыс. т в год).
Таким образом, второе направление переработки органических материалов в синтез-газ уже в настоящее время становится рентабельным и развивается, особенно в металлургии, которая постепенно переходит к бескоксовым способам производства металла, а синтез-газ в будущем станет основным восстановителем.
ПХ-переработка неорганического сырья.
Наиболее масштабные плазменные процессы реализованы в металлургии, причем на всех ее стадиях: вскрытие рудного сырья, восстановление металлов, финишная обработка полученных продуктов.
Плазменные процессы вскрытия рудных минералов. Под вскрытием подразумевается разложение химически сложного минерала на более простые вещества, из которых легко выделить необходимые продукты.
В качестве примера отметим плазменную переработку минерала циркона ZrSiO4. Схемы ПХ-реакторов для этого процесса, которые, в общем, являются типичными для большинства процессов плазменной переработки неорганического сырья, приведены на рис. 7 [7]. Измельченное исходное сырье подается в плазменную струю, нагревается до высоких температур, испытывает превращения и падает вниз, где охлаждается и проходит дальнейшую обработку.
В случае циркона, после его прохождения через плазму, образуется механическая смесь окислов циркония и кремния (ZrO2+ SiO2), которая легко разделяется.
На этом принципе в США организовано промышленное производство окиси циркония. Получаемая таким образом ZrO2 оказывается более высокого качества, чем в обычной технологии, при более низких капиталовложениях и удельных расходах электроэнергии (2.2 кВт-ч/кг против 9.2 кВт-ч/кг в обычных печах). Данный процесс характеризуется отсутствием загрязнения окружающей среды отходящими газами и побочными продуктами.
Рис. 7. ПХ-реакторы для переработки циркона фирмы «Ионарк смелтерс»:
а – с графитовыми электродами; б – с электродами, продуваемыми газом; в – с фазными плазмотронами-анодами; 1 – катод; 2 – сопло-анод; 3 – графитовые электроды; 4 – бункер с измельченным сырьем; 5 – плазмообразующий газ; 6 – фазные плазмотроны-аноды
а)
б)
в)
Аналогичным образом обрабатывают роданит, серпентиновую руду, ильменит, молибденит и другие минералы с целью получения оксидов металлов с дальнейшим их восстановлением до металлов.
ПХ-процессы в восстановительной металлургии.
Известно, что широко распространенный традиционный доменный способ получения металла, несмотря на большую производительность и высокую степень освоения, обладает рядом недостатков: многостадийностью, связанной с предварительной стадией переработки сырья (агломерация, окомковывание) и восстановителя (коксохимическое производство); загрязнением окружающей среды; материалоемкостью и энергоемкостью. В результате металлургия входит в число наиболее экологически «грязных» производств.
Поэтому в мире всегда проявлялся интерес к поискам бескоксовой и безагломерационной металлургии. Особенно этот интерес усилился в последние 10–15 лет. Это связано с истощением запасов качественных коксующихся углей и вынужденным переходом на некачественные угли, с увеличением стоимости кокса, ужесточением требований по охране окружающей среды, постоянно растущими требованиями к качеству металлов, с возможностью организации эффективного маломасштабного производства металла бескоксовым способом на базе местных ресурсов сырья и топлива при ограниченных финансовых возможностях.
В настоящее время происходит постоянное наращивание мощностей бескоксовой металлургии (удвоение производства каждые 7–8 лет), связанной в основном с бескоксовым получением в шахтных печах металлизированного сырья (губчатое железо).
Среди разрабатываемых технологий бескоксовой металлургии перспективными являются технологии плазменного восстановления. Первыми плазменными процессами, разработанными в экстрактивной металлургии, были процессы производства цветных и редких металлов. Большое внимание во многих странах уделяют разработке ПХ-процессов в черной металлургии, что связано с устранением присущих им недостатков, отмеченных выше.
Плазменное восстановление железорудного сырья разрабатывается в двух направлениях: твердофазное получение губчатого или порошкового железа и восстановительная плавка.
Типичная схема данных технологических процессов приведена на рис. 8 [14]. Здесь представлена схема процесса, разработанная фирмой SKF (Швеция). В качестве сырья для восстановительного газа могут использоваться любые органические материалы (см. рис. 6).
Интенсивно ведутся работы по плазменной восстановительной плавке чугуна из рудного сырья. Такие опытно-экспериментальные установки с противоточной шахтой, прямоточными вертикальными реакторами с водоохлаждаемыми или футерованными металлическими стенками, установленными над ванной-сборником расплава, имеются в США, Франции, Швеции, Австрии, Бельгии и ряде других стран. Получающийся чугун по составу близок к доменному.
Производство цветных и редких металлов и их соединений
Получение цветных, редких металлов и восстановительный синтез соединений их с углеродом, азотом, бором, кремнием предоставляют намного более широкие возможности для применения низкотемпературной плазмы, чем черная металлургия. Это обусловлено не только обширной номенклатурой видов сырья и продуктов, но и меньшими объемами производства, а также повышенными требованиями к его экологической чистоте.
Все варианты существующих восстановительных плазменных агрегатов в этой области можно разделить на две основные группы: в первой процессы идут с жидкой ванной расплава, во второй формирование целевого продукта осуществляется в газодисперсном потоке, при смешении его с плазменной струей. Сырье в обоих случаях может быть введено в высокотемпературную зону аппарата в виде паров, расплава, раствора или порошка необходимой крупности.
Рис. 8. Схема процесса «Плазмаред»:
1 – восстановительная печь; 2 – компрессоры; 3 – плазменный генератор восстановительного газа; 4 – смеситель газов; 5 – доломитовый фильтр
Ввод железорудного сырья
Железо
Плазменные печи с керамической ванной обладают достаточной универсальностью, и поэтому многие из них, опробованные в черной металлургии, с успехом используются для получения цветных металлов, таких как никель, кобальт, магний, ванадий, молибден, олово и др.
Для многотоннажных процессов цветной металлургии значительный интерес представляет проблема переработки рудного трудновскрываемого сырья и промышленных отходов с выделением таких металлов, как Zn, Pb, Cu, Ni, Co, Sn, Cd, Bi, РМЗ, драгметаллы, платиноиды. Некоторые из таких процессов реализованы.
Третьей стадией использования плазмы в металлургии является заключительная (финишная) обработка продуктов: переплав металлов и сплавов, их рафинирование, легирование и др. Следует отметить, что в данных промышленных процессах роль плазмы наиболее значительна. Плазменный переплав по сравнению с другими способами получения чистых металлов и сплавов обладает рядом технологических преимуществ. Это, прежде всего, возможность широкого регулирования состава и давления газовой атмосферы над расплавом, скоростей плавления заготовок и кристаллизации слитка, выбора различных рафинирующих флюсов. В зависимости от состава газовой смеси при плазменном переплаве могут проходить либо процессы рафинирования, либо легирования. Например, переплав в восстановительной атмосфере позволяет эффективно удалять кислород, а в атмосфере азота или углеводородов проводить легирование расплава азотом, углеродом или водородом [14].
Среди плазменных процессов в металлургии особое место занимают процессы получения и формообразования дисперсных материалов: плазменная порошковая металлургия – один из наиболее перспективных путей применения плазмы. Это направление охватывает получение порошков (в том числе ультрадисперсных) плазмохимическим способом (путем восстановления и синтеза), испарением, переконденсацией, диспергированием расплавов, а также процессы их формообразования, сфероидации и плакирования. Последний процесс представляет большие возможности для создания композитных материалов.
Наиболее освоенными являются процессы получения высокодисперсных порошков металлов и их соединений: карбидов, нитридов, карбонитридов, боридов, силицидов при струйно-плазменном восстановлении оксидного и галоидного сырья. В лабораторном и промышленном масштабах освоены процессы производства высокодисперсных порошков W, Wo, Ta, Nb, карбидов Ta, Nb, Ti, Si, B, W, V, Zr, Hf, нитридов Ti, Si, Nb, Ta, B, V, карбонитридов, боридов, силицидов и других соединений при восстановлении оксидов, хлоридов, фторидов и других солей этих элементов при контакте с водородом, природным газом, аммиаком, азотом и при использовании дуговых, высокочастотных и сверхвысокочастотных генераторов низкотемпературной плазмы.
Нетрадиционные направления ПХ-переработки органических материалов. К нетрадиционным способам переработки относят методы, не направленные на получение синтез-газа и ацетилена как основных продуктов. Среди них следует отметить методы переработки возобновляемого органического сырья растительного происхождения и связанных с ним продуктов и отходов, в частности продуктов и отходов лесопромышленного комплекса.
Большой мировой проблемой является утилизация и регенерация отходов целлюлозно-бумажной промышленности. Для ее решения могут быть использованы плазменные технологии.
Так, в конце 80-х годов шведские фирмы SKF steel и Stora Kopparberg в ведущих промышленных странах запатентовали совместный способ регенерации варочных химикатов из сульфатного щелока, схема которого приведена на рис. 8 [17].
В ПХ-реакторе при Т = 1250 оС сульфатный щелок разлагается на смесь Na2S, NaOH, H2, CO, H2O, CO2 и т.д. Сера находится, в основном, в соединении Na2S, но часть и в H2S; натрий содержится в Na2S, в атомарном газообразном состоянии и в NaOH (газ).
В процессе закалки происходят следующие реакции:
NaOH → NaOH (плав);
Na + H2O → NaOH (плав) + 0.5Н2;
2NaOH + CO2 → Na2CO3 (плав) + H2O;
2Na + CO2 + H2O → Na2CO3 (плав) + H2.
Образование NaOH (плав) по реакциям 1 и 2 проходит быстрее, чем реакции образования Na2CO3 (плав) по реакциям 3 и 4, поскольку реакции 1, 2 являются бимолекулярными, а реакции 3, 4 – 3-х и 4-х молекулярными. При соответствующей скорости закалки реакции 1 и 2 успевают пройти, а 3 и 4 – нет.
В результате такого процесса в плаве внизу закалочного узла будет образовываться белый щелок, а Na2CO3 будет мало. Таким образом, при реализации данного процесса при производстве целлюлозы отпадает необходимость в организации традиционных для ЦБК металлоемких, энергозатратных и экологически грязных отделов каустизации и регенерации извести.
Резюмируя вышесказанное, можно сделать вывод, что перед традиционными процессами и технологиями ПХ-процессы переработки имеют следующие преимущества:
– высокие температуры и скорости процессов приводят к значительному увеличению производительности оборудования вплоть до тысяч раз;
– ПХ-процессы в большинстве случаев являются малостадийными, не требуют промежуточных обработок, соответствующих реагентов и их производств;
– ПХ-технологии менее чувствительны к составу сырья и могут работать на менее дефицитном сырье, включая отходы производств;
– с помощью ПХ-технологий можно более полно и глубоко переработать сырье, что важно для создания малоотходных и ресурсосберегающих технологий (например, для получения 1 т ацетилена при низкотемпературном окислительном пиролизе требуется 4–5 т нефти, а по ПХ-технологии – около 2 т);
H2, CO, H2O, CO2, (H2S)
Узел
закалки
Белый щелок
Na2S, NaOH,
(Na2CO3)
ПХ-реактор
Кислород
Щелок
Плазмотрон
Рис. 8. Схема ПХ-переработки сульфатного
щелока
– ПХ-процессы хорошо моделируются, управляются и автоматизируются.
Самым большим недостатком ПХ-технологий является потребление больших количеств электроэнергии, что существенно сдерживает их широкое применение и особенно при производстве многотоннажных продуктов.
Однако темпы работ по исследованиям и разработкам ПХ-процессов и технологий с течением времени не уменьшаются, а возрастают и связано это со следующей тенденцией в развитии цивилизации: уже несколько десятилетий в мировой экономике наблюдается устойчивая закономерность опережения роста стоимости природного сырья по отношению к росту стоимости электроэнергии.
Считается, что данная ситуация будет иметь место и в будущем, а это значит, что роль ПХ-технологий возрастает и наступит момент, когда они будут достаточно рентабельны и в производствах многотоннажных продуктов для человечества. Эти и предопределяет перспективность ПХ-технологий.
2. Термодинамика и кинетика плазмохимических процессов
Предварительным этапом проектирования ПХ-технологий и соответствующего оборудования является оценка характеристик и параметров плазмохимической среды как определяющего агента ПХ-процесса.
2.1 Термодинамический анализ плазмохимических систем
Почти во всех случаях такие оценки для ПХ-технологий можно получить из термодинамического анализа ПХ-систем. Для равновесных (квазиравновесных) ПХ-процессов ТД-расчеты являются необходимым этапом проектирования. Для неравновесных ПХ-процессов ТД-расчеты позволяют оценить предельные значения различных параметров плазмохимических процессов. Методы ТД-анализа ПХ-систем подробно приводятся в работах [15, 18].
2.2 Кинетика плазмохимических процессов
Из термодинамики ПХ-процессов нельзя получить информацию об их временных характеристиках (скоростях превращений одних компонентов в другие и т.д.), которые зачастую являются определяющими при разработке ПХ-технологий и устройств для их осуществления.
Поэтому наряду с термодинамическими расчетами при разработке технологий стараются по возможности провести кинетический анализ происходящих процессов.
Химическая кинетика изучает закономерности протекания химических реакций во времени. Она позволяет установить изменение состава реагирующих компонентов во времени от условий протекания процесса (температуры, исходного состава сырья и т.д.). С помощью кинетических данных можно найти время образования максимального количества целевых продуктов и соответствующие размеры реакционной аппаратуры.
В зависимости от состояния плазменные среды можно условно разделить на равновесные (квазиравновесные) и неравновесные [15, 19].
2.2.1 Кинетика равновесных процессов
В равновесных условиях изотермическую плазму можно рассматривать как обычную высокотемпературную газовую систему, для описания которой используется классическая химическая кинетика.
В основе классической химической кинетики лежит представление о том, что химические взаимодействия осуществляются при термодинамически равновесных распределениях энергии по непрерывным (распределение Максвелла) и дискретным (распределение Больцмана) энергетическим уровнями, и такая ситуация не меняется в течение всего химического взаимодействия.
Данная ситуация реализуется, когда скорости физических процессов установления равновесия по поступательным, вращательным и колебательным степеням свободы существенно превышают скорости химических процессов. Например, при температурах Т = 3·103÷1.5·104 и атмосферном давлении, что соответствует наиболее распространенному виду квазинейтральной плазмы дугового разряда, длительности физических процессов характеризуются следующими величинами:
– среднее время молекулярного взаимодействия – 10-14с;
– среднее время свободного пробега молекул, совпадающее по порядку со временем установления распределения по поступательным и вращательным степеням свободы – 10-9с;
– время колебательной релаксации – 10-7с;
– характерное время ПХ-реакций – 10-4 ÷ 10-3.
При низких давлениях эти величины существенно меняются, что приводит к неравновесию.
Ограничимся рассмотрением только гомогенных процессов, происходящих в газовой среде.
В химической кинетике реакцию превращения исходных веществ A1, A2, …, An в конечные продукты B1, B2, …, Bl принято записывать в общем виде следующим уравнением:
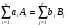 , (1)
, (1)
где 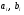 – стехиометрические коэффициенты, выражаемые числами, пропорциональными количествам молей реагирующих компонентов.
– стехиометрические коэффициенты, выражаемые числами, пропорциональными количествам молей реагирующих компонентов.
Любая химическая реакция обратима, то есть может идти как в прямом, так и обратном направлении.
Важной количественной характеристикой химической реакции является ее скорость, которая определяется изменением концентраций реагирующих веществ (или продуктов реакций) во времени.
Таким образом, скоростью реакции по некоторому компоненту i называется изменение количества этого компонента за единицу времени в единице объема:
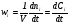 , (2)
, (2)
где ni – число молей компонента i в объеме V, 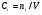 – его мольная концентрация.
– его мольная концентрация.
Зависимость скорости реакции от концентраций реагентов определяется законом действующих масс: скорость химической реакции пропорциональна произведению концентраций реагирующих веществ, взятых в степенях, соответствующих их стехиометрическим коэффици-ентам:
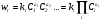 , (3)
, (3)
где ki – константа скорости i реакции.
Химические процессы совершаются при столкновении реагирующих частиц. В зависимости от числа молекул, участвующих в элементарном акте столкновения, и их превращений химические реакции характеризуются молекулярностью и порядком реакции.
Порядок реакции определяется числом частиц, одновременно участвующих в элементарном акте химической реакции, и равен сумме стехиометрических коэффициентов реакции 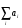 . Различают реакции первого, второго, третьего порядка, соответствующие значениям этой суммы, равной 1, 2, 3. На практике порядок реакции определяют по зависимости скорости реакции от концентрации каждого из реагирующих компонентов по формуле (3).
. Различают реакции первого, второго, третьего порядка, соответствующие значениям этой суммы, равной 1, 2, 3. На практике порядок реакции определяют по зависимости скорости реакции от концентрации каждого из реагирующих компонентов по формуле (3).
Молекулярность же определяется числом молекул, участвующих в элементарном акте столкновения и испытавших химическое превращение в данном акте. На практике приходится иметь дело только с тремя типами реакций: моно-, би- и тримолекулярными реакциями. Реакции, в которых принимают участие более трех частиц, протекают не в один акт, а состоят из нескольких элементарных актов (стадий) и называются сложными реакциями.
Единицы измерения константы скорости реакции определяются с использованием формулы (3) и зависят от единиц, в которых выражены концентрации реагентов, а также от порядка реакции (например, 1/с, м3/(моль·с), м6/(моль2·с) для реакции 1, 2 и 3-го порядков соответственно).
Следует отметить, что часто порядок реакции, полученный экспериментально, не совпадает с количеством молекул, участвующих в реакции, и может быть даже дробной величиной. Это несовпадение сви-детельствует о сложном механизме реакции, протекающей в несколько стадий.
В качестве примера рассмотрим достаточно важную для человечества реакцию окисления азота. Брутто-формула данной реакции имеет вид (второй порядок реакции): N2 + O2 2N, а выражение для ее скорости – 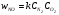 .
.
Однако экспериментальные результаты показали, что данная зависимость для скорости оказывается неверной. Детальное исследование данной реакции показало [20], что процесс получения оксида азота протекает в несколько элементарных стадий:
O2 + M O + O + M;
O + N2 N + NO,
где под частицей М подразумевается любая частица смеси (O2, N2, N, O). Первая реакция (как прямая, так и обратная) протекает с высокой скоростью, и концентрацию атомарного кислорода можно считать равновесной. Тогда, используя закон действующих масс в состоянии ТД-равновесия (см. ниже), можно получить выражение для концентрации атомарного кислорода: 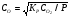 , где КР – константа равновесия диссоциации О2.
, где КР – константа равновесия диссоциации О2.
Вторая реакция является достаточно медленной, и именно она определяет результирующую скорость окисления азота. По закону действующих масс скорость второй реакции будет выражаться формулой: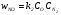 , где k2 – константа скорости второй реакции. Подставляя сюда значение для СО, получим результирующую скорость окисления азота, порядок которой оказывается дробным (3/2):
, где k2 – константа скорости второй реакции. Подставляя сюда значение для СО, получим результирующую скорость окисления азота, порядок которой оказывается дробным (3/2):
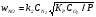 .
.
Механизмом химической реакции называют последовательность элементарных реакций (стадий), протекающих в реагирующей системе. Механизмы реакций могут быть весьма разветвленными и включать в себя множество реакций, не входящих в уравнение брутто-реакции, связывающей исходные вещества и конечные продукты реакции. Сложные механизмы химических реакций характерны для ПХ-процессов, происходящих при высоких температурах.
В классической химической кинетике константа скорости задается формулой Аррениуса:
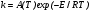 , (4)
, (4)
где Е – энергия активации.
Теоретическое определение константы скорости реакции связано со значительными трудностями, особенно при высоких температурах. Поэтому, в основном, k определяется экспериментально.
Если данный компонент i участвует в нескольких реакциях, то суммарную скорость его изменения, с учетом обратных реакций, можно записать следующим образом:
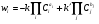 , (5)
, (5)
где k' – константа скорости обратной реакции.
При прохождении достаточно большого времени от начала реакций скорости прямых и обратных реакций уравновешиваются, и наступает состояние равновесия, когда количества исходных веществ и конечных продуктов не изменяются во времени, а суммарная скорость будет равна нулю (wi = 0).
Из (5) следует, что в состоянии равновесия при wi = 0 отношение произведений концентраций исходных веществ и конечных продуктов постоянно. Величина этого отношения носит название константы равновесия, а само выражение – закона действующих масс для ТД-равно-весия:
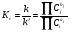 . (6)
. (6)
Значение Кс можно достаточно точно рассчитать термодинамическими методами. В справочной литературе имеется много данных по Кс
для различных реакций. Константа равновесия имеет большое вспомогательное значение в химической кинетике, поскольку, зная эту величину, из (6) можно легко вычислить константу скорости обратной реакции k', если известна k.
Очень часто в справочной литературе константа равновесия выражается через парциальные давления реагирующих веществ:
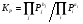 . (7)
. (7)
Связь между Кс и КР легко найти, используя уравнение состояния идеального газа 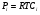 :
:
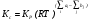 . (8)
. (8)
В статических условиях система для расчета кинетики процесса, в основном, состоит из уравнений кинетики, которые в общем виде можно записать следующим образом:
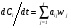 , (9)
, (9)
где J – число элементарных реакций; aij – стехиометрический коэффициент, с которым сам i-й компонент входит в уравнение j-й реакции (берется со знаком плюс, если i-й компонент является продуктом реакции, и со знаком минус, если он является исходным веществом). Если же этот компонент вообще не принимает участия в j-й реакции, то aij = 0.
С учетом (3) уравнения (9) приобретут следующий вид:
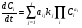 . (10)
. (10)
Эти уравнения справедливы для реакций, идущих при постоянном объеме реагирующей системы. Если процесс протекает в неизотермических условиях с изменением объема реагирующей системы, как в результате изменения температуры, так и в результате изменения количества молей реагирующих компонентов, то, используя определение концентрации , уравнения (10) можно легко привести к виду:
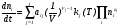 , (11)
, (11)
где 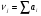 – порядок j-й реакции.
– порядок j-й реакции.
Совокупность таких уравнений, составленных в соответствии с принятым механизмом химических реакций для всех реагирующих компонентов системы (исходные вещества, промежуточные и конечные продукты), представляют собой систему обыкновенных дифференциальных уравнений, описывающих кинетику химических превращений ПХ-про-цесса.
Число таких уравнений, которые можно составить для реагирующей системы, равно количеству реагирующих компонентов. Однако не все они являются независимыми. Если в системе присутствует a различных химических элементов и s реагирующих компонентов, то количество линейно-независимых кинетических уравнений будет (s – a). Это дает возможность сократить число дифференциальных уравнений в системе, заменив некоторые из них алгебраическими уравнениями материального баланса:
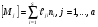 , (12)
, (12)
где [Mj] – суммарное количество молей j-го химического элемента, 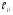 – число атомов j-го элемента в молекуле компонента i, ni – число молей компонента i.
– число атомов j-го элемента в молекуле компонента i, ni – число молей компонента i.
Записанная таким образом система нелинейных обыкновенных дифференциальных уравнений (11), (12), как правило, не имеет решения в аналитической форме и решается численными методами. В качестве начальных условий используются начальные концентрации исходных веществ. Концентрации промежуточных веществ и продуктов в начале реакции обычно равны нулю.
В результате интегрирования данной системы уравнений получают зависимости изменения состава реагирующих компонентов во времени, называемые кинетическими кривыми.
ПХ-процессы могут протекать в неизотермических условиях. Температура реагирующих веществ в плазменном процессе может меняться за счет тепловых эффектов реакций, а также в результате подвода энергии к системе от электрического разряда или плазменной струи и отвода энергии теплообменом.
Поэтому для расчета характеристик ПХ-системы в данном случае к вышеприведенной системе добавляют еще уравнение состояния газа
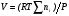 , (13)
, (13)
а также уравнение энергии (уравнение теплового баланса)
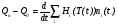 , (14)
, (14)
где Qn(t), Q0(t) – мощности подводимой и отводимой энергии в ходе процесса; ni, Hi(T) – количество молей и полная мольная энтальпия компонента i.
Особенности решения системы кинетических уравнений
Следует отметить некоторые особенности, возникающие при решении системы кинетических уравнений.
Во-первых, в ряде случаев значения констант скоростей элементарных реакций ПХ-процесса определяются с точностью, не превышающей 1–2-х порядков величины. Поэтому в данном случае необходимы дополнительные исследования на устойчивость решения данной системы к ошибкам в значениях констант скоростей химических реакций.
Поэтому при решении системы более корректно применение «статистического» подхода. В нем используется не точка в пространстве констант скоростей, а некоторая область, обусловленная их погрешностями, что, в свою очередь, приводит к расчету не кинетической кривой, а некоторой области в пространстве концентрация – время. Если эти области недостаточно узки, то решать систему с такими погрешностями в значениях констант скоростей не имеет смысла.
Во-вторых, очень часто в реагирующей системе одновременно протекают реакции с сильно различающимися скоростями (быстрые и медленные реакции). Данная ситуация вносит существенные математические трудности для численного решения системы уравнений химической кинетики.
При математическом моделировании кинетики ПХ-процессов необходимо численно решать задачу Коши для системы нелинейных обыкновенных дифференциальных уравнений (ОДУ):
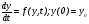 ,
,
где y – вектор переменных; у0 – вектор начальных значений (при t = 0).
При этом якобиан системы (матрица 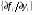 ) имеет сильно различающиеся между собой действительные части собственных значений (систему уравнений называют жесткой). Свойство жесткости системы кинетических уравнений и является отражением того факта, что система ОДУ описывает протекание процессов с сильно различающимися характерными временами (быстрые и медленные реакции).
) имеет сильно различающиеся между собой действительные части собственных значений (систему уравнений называют жесткой). Свойство жесткости системы кинетических уравнений и является отражением того факта, что система ОДУ описывает протекание процессов с сильно различающимися характерными временами (быстрые и медленные реакции).
При использовании классических разностных методов Рунге –Кутта, Адамса и др. для численного решения систем жестких ОДУ условие устойчивости решения требует чрезмерного уменьшения шага интегрирования и приводит к большим временам счета и накоплению глобальных ошибок. В связи с чем для решения жестких ОДУ был разработан ряд специальных численных методов (например, методы Гира и их модификации), в которых, как правило, на разных временных интервалах используются разные классические методы (пакеты программ GEAR, EPISODE, LSODE и др.) [15].
Основной проблемой при кинетических расчетах сложных реакций является отсутствие необходимой информации о константах скоростей химических реакций. Знание общих свойств элементарных реакций (стадий) может существенно облегчить нахождение данных констант, поэтому кратко охарактеризуем некоторые реакции.
Свойства элементарных реакций ПХ-процессов.
ПХ-процессы, как правило, характеризуются протеканием сложных реакций, представляющих собой совокупность простых химических реакций или элементарных стадий, следующих одна за другой или совершающихся параллельно. Каждую из этих стадий можно рассматривать как самостоятельную химическую реакцию со своими исходными веществами и продуктами. При этом исходными веществами и продуктами реакций могут выступать не только стабильные, но и лабильные промежуточные частицы – атомы, ионы, свободные радикалы.
Химические реакции, состоящие из одной стадии, то есть осуществляющиеся прямым переходом реагирующих частиц в продукты реакции, называются элементарными.
Для ПХ-процессов характерными элементарными реакциями являются реакции диссоциации. Диссоциация молекул на атомы и радикалы является одним из основных химических процессов, протекающих при высоких температурах, поэтому сведения о кинетике распада представляют значительный интерес для ПХ-реакций.
Согласно существующим представлениям, диссоциация молекул состоит из нескольких стадий. На первой стадии происходит активация молекул при столкновениях, когда молекула получает энергию, превышающую энергию активации Е:
 . (15)
. (15)
На 2-й стадии происходят два конкурирующих процесса: дезактивация молекулы при столкновении:
 (16)
(16)
и распад молекулы:
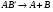 . (17)
. (17)
В целом диссоциация относится к мономолекулярным реакциям, поскольку химическому превращению подвергается одна молекула, и может иметь 1-й или 2-й порядок в зависимости от условий протекания процесса: давления, строения молекулы, числа атомов, образующих молекулу. Такое изменение порядка соответствует смене лимитирующей стадии процесса.
Допустим, что время между столкновениями молекул велико (низкое давление) по сравнению со временем, необходимым для распада возбужденной молекулы. В результате большинство возбужденных молекул распадается раньше, чем новые столкновения приведут к их дезактивации. Очевидно, что в этом случае скорость реакции определяется скоростью 1-й стадии (15), имеющей второй порядок.
Если же время между столкновениями молекул мало по сравнению со временем распада (высокое давление), то скорость суммарной реакции диссоциации будет определяться скоростью распада возбужденных молекул (17), а реакция диссоциации здесь имеет 1-й порядок.
Кроме того, на скорость лимитирующей стадии распада, а следовательно, и на порядок реакции диссоциации влияет строение молекул. В зависимости от числа атомов, входящих в состав молекулы, для каждого вида молекул существует определенный предел давления, при котором меняется порядок реакции.
Как правило, при P > 1 атм для простых молекул, содержащих не более 4 атомов, процесс распада лимитируется скоростью накопления внутренней энергии, необходимой для разрыва связей между атомами (скорость распада здесь велика). Поскольку такое накопление происходит при столкновениях по реакции (15), то диссоциация должна иметь 2-й порядок. Это относится к диссоциации 2-атомных (например, N2, O2, H2 и др.) и 3-атомных (например, O3, NO2, N2O и др.) молекул. 4-атомные молекулы (например, Fe2O2, NO2Cl) обычно распадаются по 2-му порядку, хотя порядок может быть и промежуточным между 1-м и 2-м.
Более сложные молекулы с числом атомов большим четырех распадаются по 1-му порядку, причем скорость суммарного процесса контролируется разрывом связей (скорости распада здесь малы).
Реакция, обратная диссоциации, называется реакцией рекомбинации (ассоциации) и включает те же элементарные процессы, только протекающие в обратном направлении. Реакция начинается с присоединения атомов и (или) радикалов по реакции: 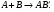 В следующей стадии должно произойти столкновение, дезактивирующее сильно возбужденную молекулу:
В следующей стадии должно произойти столкновение, дезактивирующее сильно возбужденную молекулу: 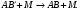
Рекомбинация относится к бимолекулярным реакциям, поскольку в химическом превращении участвуют 2 частицы, и может иметь различный порядок, который зависит от условий протекания процесса и определяется порядком соответствующей реакции диссоциации. Порядок реакции рекомбинации на единицу больше соответствующей реакции диссоциации. Так, если диссоциация протекает по 1-му порядку, то рекомбинация – по 2-му, если диссоциация протекает по 2-му, то рекомбинация – по 3-му.
Например, реакция диссоциации и рекомбинации Н2 при атмосферном давлении и высоких Т определяется следующим выражением:
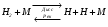 .
.
Это связано с тем, что при образовании простой молекулы из атомов или небольших радикалов для отвода избыточной энергии от активированной молекулы необходимо столкновение с какой-нибудь 3-й частицей. В противном случае рекомбинация не произойдет и столкнувшиеся атомы разлетятся снова. Отсюда становится ясным, почему указанная реакция является реакцией 3-го порядка.
При рекомбинации больших молекул и сложных радикалов столкновение с 3-й частицей необязательно, так как избыточная энергия вновь образующейся связи может перераспределяться по другим связям, которых в сложной молекуле достаточно много. Например, рекомбинация радикалов СН2 протекает по 2-му порядку: 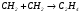 .
.
Большую роль в ПХ-процессах играют реакции рекомбинации атомов и небольших радикалов, которые образуются в значительных количествах при высоких Т. Эти реакции, как правило, протекают по 3-му порядку и имеют определенные особенности. Во-первых, они имеют нулевую энергию активации; во-вторых – отрицательную зависимость констант скоростей от Т (таблица 1).
Таблица 1
-
Реакции
Т, К
k, м6/(кмоль2·с)
Н + Н + М Н2 + М
300
1700
3500
1010.1
109.5
109.3
О + О + М О2 + М
300
2200
4000
109.2
108.8
108.1
N + N + M N2 + M
300
109.5
H + OH + M H2O + M
1900
109.6
Следует отметить, что в большинстве случаев константы скоростей лежат вблизи значений 109.5 0.5 м6/(кмоль2·с) при 300 К, и примерно на порядок ниже – при 3000 К. Поэтому приведенные данные используются для оценки констант скоростей реакций рекомбинаций любых атомов.
Рекомбинация более сложных радикалов протекает, как правило, по 2-му порядку, имеет нулевую энергию активации и константу скорости, слабо зависящую от температуры (k ~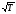 ). Например, для реакции CH3 + CH3 C2H6
). Например, для реакции CH3 + CH3 C2H6 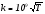 м3/(кмоль·с).
м3/(кмоль·с).
Наряду с реакциями диссоциации и рекомбинации в ПХ-процессах проходят обменные реакции. Эти реакции отличаются сложностью, так как могут протекать несколькими путями с образованием различных продуктов. Например, взаимодействие атомов кислорода с моноксидом азота может осуществляться 2-мя путями:
O + NO + M NO2 + M,
O + NO O2 + N.
Тепловой эффект Q, равный разности энергий образуемой и разрываемой связей, для реакций этого типа может быть как положительным, так и отрицательным. Как показал Н. Н. Семенов, для большинства реакций этого типа энергию активации можно приближенно определить для экзотермического процесса по формуле: Еэкз = 48.2 – 1.05·Q (кДж/моль); для эндотермического Еэнд = 48.2 – 3.14·Q (кДж/моль).
Эти формулы позволяют оценить энергию активации реакции передачи атома от молекулы к свободному радикалу или атому, если известны энергии разрываемой и образуемой связей (то есть Q).
Величины предэкспоненциальных множителей обменных реакций, в основном, определяются степенью сложности реагирующих частиц и изменяются от 107 до 1010 м3/(кмоль·с). Ввиду такой неопределенности для нахождения констант скоростей обменных реакций желательно использовать экспериментальные данные.
Строгое рассмотрение элементарных химических реакций и расчет констант скоростей должны основываться на методах квантовой механики. Однако пока квантовомеханическое описание химических реакций разработано недостаточно, что объясняется сложностью процессов, происходящих при соударениях частиц – реагентов.
2.2.2 Неравновесные плазмохимические процессы
Плазмохимические системы являются открытыми системами, существующими, пока имеется поток энергии через них. Этот факт является фундаментальной причиной их неравновесности.
Кинетика химических превращений определяется динамикой соударения частиц и статистикой поведения их ансамблей. Различия в динамике соударения частиц и свойствах взаимодействующих ансамблей в классических системах, далеких от состояния равновесия, позволяют выделить следующие основные особенности кинетики неравновесных процессов:
– нельзя отделить химическую кинетику от физической (установление распределения по энергетическим уровням), а сами химические процессы вызывают существенное отклонение системы от равновесия;
– любая ПХ-реакция является многоканальным процессом, происходящим с участием целого ряда возбужденных состояний;
– многоканальность процесса со своей пороговой энергией каждого канала приводит к невозможности описания процесса в рамках представлений о единой энергии активации типа аррениусовой.
Одновременный учет физической и химической кинетики при описании неравновесных процессов можно провести с помощью уравнения Паули, связывающего вероятности нахождения системы в различных квантовых состояниях (Pi, Pk) с вероятностями перехода между состояниями (aik, aki), отнесенными к единице времени:

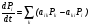 . (18)
. (18)
Уравнение Паули есть уравнение баланса, в котором должны учитываться переходы между квантовыми состояниями частиц, вступление частиц в этих состояниях в химические реакции, а также возможность генерации возбужденных частиц под действием внешних по отношению к данной системе источников («накачка»).
Для простейшего случая мономолекулярной реакции, протекающей в термостате инертного газа, концентрация частиц которого является постоянной, уравнение Паули примет вид:
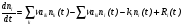 , (19)
, (19)
где ni – концентрация реагирующих молекул в i-м квантовом состоянии;
– частота столкновений частиц; aik, aki – рассчитанные на одно столкновение вероятности того, что при столкновении реагирующих молекул с молекулами термостата первые переходят из k-го в i-е состояние и наоборот; ki – константа (коэффициент) скорости химической реакции молекул в i-м состоянии; Ri(t) – скорость «накачки» i-го состояния.
Если не учитывать переходы между квантовыми состояниями молекул, уравнение Паули сводится к обычным уравнениям химической кинетики.
Уровневые константы скорости реакции.
Так как ПХ-процесс всегда многоканален, статистические константы (коэффициенты) скорости реакции следует определять как константу средней скорости по всевозможным каналам. Усреднение должно проводиться по скоростям и квантовым состояниям реагирующих частиц. Это значит, что в выражение для константы скорости реакции должны входить в явном виде функции распределения компонентов.
Константы (коэффициенты) скоростей взаимодействия частиц в фиксированных квантовых состояниях называются уровневыми константами (коэффициентами) скоростей реакций.
Учитывая это, запишем скорость реакции по компоненту i для бимолекулярной реакции 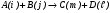 :
:
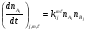 . (20)
. (20)
Здесь 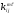 – уровневый коэффициент скорости реакции частицы А в i-м состоянии с частицей В в j-м состоянии с образованием продуктов в состояниях m и
– уровневый коэффициент скорости реакции частицы А в i-м состоянии с частицей В в j-м состоянии с образованием продуктов в состояниях m и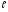 .
.
Найдем выражение для уровневого коэффициента скорости реакции. Обозначим через 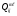 сечение взаимодействия частиц A(i) и B(j), приводящее к появлению продуктов C(m) и D(), а через
сечение взаимодействия частиц A(i) и B(j), приводящее к появлению продуктов C(m) и D(), а через 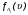 и
и 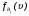 функции распределения частиц A(i) и B(j) по скоростям соответственно.
функции распределения частиц A(i) и B(j) по скоростям соответственно.
За единицу времени одна частица 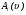 столкнется (с химическим превращением) со всеми частицами
столкнется (с химическим превращением) со всеми частицами 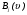 , находящимися в объеме
, находящимися в объеме 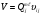 , где
, где 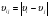 – относительная скорость сталкивающихся частиц A(i) и B(j).
– относительная скорость сталкивающихся частиц A(i) и B(j).
Количество частиц (в объеме V), имеющих скорости в интервале 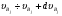 , будет определяться выражением:
, будет определяться выражением: 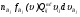 . Концентрация частиц , имеющих скорости в интервале
. Концентрация частиц , имеющих скорости в интервале 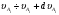 , будет равна
, будет равна 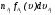 .
.
Таким образом, общее число столкновений (с химическим превращением) в единицу времени частиц Ai, находящихся в единице объема (то есть скорость реакции), можно записать следующим образом:
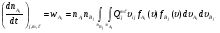 (21)
(21)
Из сравнения этого выражения с обычным выражением закона действующих масс (3) следует, что уровневый коэффициент скорости реакции можно записать следующим выражением:
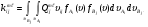 (22)
(22)
Усредненный коэффициент для такой неравновесной многоканальной реакции можно определить, просуммировав (20) по всем начальным (i, j) и конечным (m, ) состояниям. В результате получим:
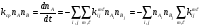 . (23)
. (23)
Отсюда легко получается искомое выражение для среднего значения константы скорости неравновесной химической реакции:
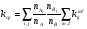 , (24)
, (24)
где nA, nB – общие концентрации компонентов А и В во всевозможных состояниях: 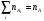 ,
, 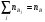 .
.
В системе, близкой к равновесию, при выполнимости распределений Максвелла и Больцмана выражение (24) с учетом (22) приводит к аррениусовой температурной зависимости суммарного коэффициента скорости процесса:
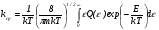 ,
,
которая для модели твердых сфер принимает аррениусовую форму:
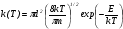 ,
,
где m, – масса и кинетическая энергия частицы.
3. Технологическое оформление плазмохимических процессов
Организация технологического процесса плазмохимической переработки материалов проводится по схеме, приведенной на рис. 4. Рассмотрим более подробно отдельные узлы этой схемы.
Одним из наиболее важных узлов данной схемы является генератор плазмы (плазмотрон). Однако физика и техника генераторов плазмы изучается в отдельном курсе, достаточно полно описана в литературе [3–7, 19] и здесь рассматриваться не будет.
Главным элементом в технологической схеме является плазмохимический реактор.
3.1 Плазмохимический реактор
ПХ-реактор, как правило, можно рассматривать состоящим из двух частей: смесителя плазмы и сырья и собственно реактора. Смеситель должен обеспечивать получение гомогенной смеси реагентов на входе в реактор при некоторой температуре, величина которой определяется исходя из кинетических и термодинамических характеристик химического процесса.
При проектировании химических реакторов важно иметь информацию о распределениях потоков газа (жидкости) в них, а также уметь определять время нахождения реагентов внутри реактора, поскольку от этого времени зависит степень превращения исходных веществ в продукты. Задача эта является не простой, поскольку в реальных реакторах, особенно сложной конструкции, реагирующие вещества движутся по сложным, трудно прогнозируемым траекториям.
В качестве одной из мер, предназначенных для решения задачи, является введение функций распределения по временам пребывания в объеме реактора.
3.1.1 Функции распределения по временам пребывания
Отметим, что при рассмотрении процессов в химических реакторах понятия жидкости и газа часто эквивалентны, поскольку они подчиняются одним и тем же закономерностям, а для их описания используется один и тот же математический аппарат.
Рассмотрим поток жидкости (газа), проходящий через реактор в условиях неизменной плотности и отсутствия химических реакций. Тогда среднее время пребывания жидкости в объеме будет определяться выражением:
= V/G, (25)
где V – объем реактора, G – объемный расход жидкости.
В дальнейшем по мере необходимости будем пользоваться понятием безразмерного времени:
= t/τ = Gt/V, (26)
где t – натуральное время.
Различные элементы жидкости («жидкие» частицы) могут проходить через реактор различными путями, и, следовательно, периоды присутствия этих элементов в объеме реактора будут неодинаковыми.
Пусть каким-то образом удается зарегистрировать распределение I – интервалов времени, в течение которых эти элементы присутствуют в реакторе, считая от некоторого момента t = 0. Тогда Idt составит долю элементов жидкости, для которых интервалы времени присутствия внутри реактора находятся в пределах от t до t + dt. Поскольку сумма всех долей жидкости в реакторе равна единице, то выполняется следующее выражение (условие нормировки функции распределения I):
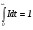 . (27)
. (27)
Очевидно, что доля содержащихся в реакторе элементов жидкости, для которых время присутствия меньше некоторого значения t1, составит величину 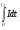 . Доля находящихся в реакторе элементов жидкости со временем присутствия больше t1 будет равна
. Доля находящихся в реакторе элементов жидкости со временем присутствия больше t1 будет равна 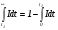 .
.
Пусть теперь удалось найти распределение Е – интервалов времени, прошедших от момента попадания данного элемента жидкости в объем реактора до момента выхода его из реактора (распределение по временам пребывания).
По аналогии с предыдущей функцией величина Edt составит долю элементов жидкости, для которых время от момента их попадания в объем реактора до появления в потоке на выходе реактора находится в диапазоне от t до t + dt. Имеет место аналогичное условие нормировки:
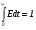 . (28)
. (28)
С использованием функции распределения по временам пребывания Е среднее время пребывания жидкости в реакторе составит величину:
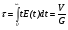 . (29)
. (29)
Функция E(t) связана с функцией I(t) соотношением:
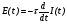 . (30)
. (30)
При использовании безразмерного времени (26) имеют место следующие соотношения:
I() = I(t); E() = E(t); 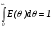 ;
; 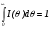 .
.
Знание указанных функций распределения, характеризующих данный реактор, необходимо для расчета степени превращения веществ в реакторе и определения его оптимального размера и конфигурации.
Например, степень превращения X исходного реагента в ходе реакции 1-го порядка dn/dt = –kn определяется следующим выражением:
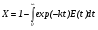 . (31)
. (31)
Для экспериментального определения функций распределения I и Е используют методы, основанные на исследовании отклика данной системы на возмущение. Возмущающее воздействие в рассматриваемом случае проточного реактора заключается во введении в поток на его входе какого-либо вещества (трассера), химически не реагирующего с веществом потока.
Сигналы возмущения могут в принципе иметь различную форму, однако чаще всего используют ступенчатое или импульсное возмущение, поскольку это существенно упрощает анализ состояния исследуемой системы.
Действительно, легко видеть, что при внесении возмущения в виде длинной ступенчатой функции (длинный импульс) отклик F системы связан с функцией распределения I жидкости по временам присутствия в объеме реактора простыми соотношениями: I = 1 – F, если отсчет вести от переднего фронта, и I = F при отсчете от заднего фронта импульса. При этом длина импульса должна быть больше времени установления стационарного распределения трассера по объему реактора (то есть значительно больше среднего времени пребывания жидкости в реакторе).
При использовании возмущающего воздействия в виде короткого импульса, длительность которого много меньше , отклик С системы совпадает с функцией Е распределения жидкости по временам пребывания, то есть С = Е.
Для удобства сравнительного анализа характеристик различных реакторов вводят понятия об идеальных реакторах.
Реактором идеального вытеснения называется реактор, в котором в любом поперечном сечении, нормальном к направлению движения потока жидкости, скорость и все свойства жидкости (давление, состав, температура и др.) распределены равномерно, а продольные диффузионные потоки отсутствуют.
Таким образом, в реакторе идеального вытеснения все элементы жидкости проходят через реактор за одинаковое время, и последовательность изменения свойств жидкости одинакова для всех элементов. Поскольку диффузия из одного «поперечного» элемента в другой отсутствует, степень превращения в каждом элементе остается одной и той же.
Это позволяет рассматривать каждый такой элемент как реактор периодического действия с длительностью цикла, равной времени прохождения элемента через реактор.
Реактор идеального перемешивания определяется как реактор, в котором поступающая в него жидкость мгновенно перемешивается с содержимым объема реактора, так что состав смеси в любой точке объема реактора одинаков.
Таким образом, реактор идеального перемешивания является по смыслу крайней противоположностью реактора идеального вытеснения.
В таблице 2 приведены функции распределения I и Е для реакторов различного типа.
Таблица 2
Реактор идеального вытеснения
Реактор идеального перемешивания
Реальный реактор
1
1
I
S = 1
1
1
I
S = 1
1
1
I
S = 1
exp (– )
1
E
S = 1
1
1
E
S = 1
exp (– )
1
E
S = 1
Видно, что функции распределения для реального реактора представляют собой нечто среднее между соответствующими функциями для реакторов идеального вытеснения и перемешивания.
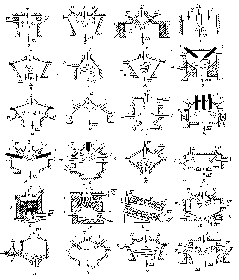
Разработано огромное количество различных конструкций ПХ-реакторов, некоторые из них приведены на рис. 9 [7].
Некоторые реальные химические реакторы по своим характеристикам оказываются достаточно близкими к одному из идеальных реакторов. Для оценки принадлежности какого-либо реактора к классу аппа-ратов идеального вытеснения или смешения следует сопоставить скорости массообмена со скоростью химической реакции.
Для этого можно воспользоваться критериями, разработанными различными авторами, например, «критерием перемешивания» Кп, предложенным Абаевым [1]:
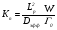 , (32)
, (32)
где Lp – длина реактора, Dэфф – эффективный коэффициент массообмена (обычно коэффициент турбулентной диффузии), W – скорость химической реакции, Г0 – исходная концентрация реагента.
При Кп << 1 режим потока в реакторе близок к режиму реактора идеального перемешивания, а при Кп >>1 – к режиму реактора идеального вытеснения.
Оценим Кп для ПХ-реактора лабораторной установки пиролиза природного газа в струе водородной плазмы, имеющей следующие характеристики: Lp = 10d = 6 см; характерное время химической реакции (Г0/W) ~ 10-4c (при Т реакции ~ 2000 K) и эффективный коэффициент турбулентной диффузии в рассматриваемых условиях Dэфф ~ 103 см2/с (исходя из результатов измерения времени перемешивания до молекулярных масштабов).
Подставляя данные результаты в (32), получаем для критерия перемешивания значение Кп 400 >>1. Отсюда следует, что данный реактор по своим характеристикам близок к реактору идеального вытеснения. Аналогичная ситуация имеет место для большинства плазмоструйных ПХ-реакторов. Поэтому в дальнейшем мы будем рассматривать только реакторы вытеснения.
Отмеченная выше близость реального и идеального реакторов не означает полной их эквивалентности. Существует ряд принципиальных и полностью неустранимых различий между идеальными и реальными реакторами вытеснения.
3.1.2 Характеристики реального реактора вытеснения
Различия в реакторах идеального и реального вытеснения объясняются следующими причинами.
Во-первых, из-за отсутствия материалов, выдерживающих плазменные температуры, стенки реального реактора приходится охлаждать и поддерживать при более низких температурах. В результате появляются радиальные градиенты температур и связанные с ними радиальные градиенты скорости потока.
Рис. 9. Конструктивные схемы плазмохимических реакторов:
1 – электроды; 2 – футеровка; 3 – соленоиды; 4 – ввод газа; 5 – фильтр; 6 – шнек; 7 – подача жидкости; 8 – диафрагма; I – струя плазмы; II – сырье; III – продукты; IV – защитный газ
Это приводит к тому, что скорости химических превращений в пристеночной области реактора значительно меньше, чем в центральных областях. При этом увеличение времени пребывания пристеночного слоя газа в реакторе, за счет снижения скорости потока вблизи охлаждаемых стенок, лишь незначительно компенсирует данный эффект.
Для того чтобы в этих условиях обеспечить расчетную степень химического превращения, необходимо увеличить длину реактора. Но это приведет к снижению селективности ПХ-процесса, целевой продукт которого является промежуточным в цепи химических превращений в реакторе, поскольку скорость превращения в высокотемпературном ядре потока остается высокой.
Для увеличения эффективности ПХ-реактора, предназначенного для проведения процесса, целевой продукт которого является промежуточным, стенку реактора теплоизолируют (футеруют) и максимально повышают ее температуру (насколько позволяет изолирующий материал и материал стенки реактора).
Второй причиной, приводящей к различию реальных и идеальных реакторов вытеснения, является влияние турбулентной диффузии в направлении оси реактора (продольная диффузия). Ее влияние тем сильнее, чем больше величина 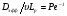 , где Ре – критерий Пекле. Для вышеприведенного примера эта величина равна 3·10-3 (при средней скорости потока 500 м/с), что значительно меньше единицы, и влиянием этого эффекта на функцию распределения по временам пребывания Е(t) можно пренебречь.
, где Ре – критерий Пекле. Для вышеприведенного примера эта величина равна 3·10-3 (при средней скорости потока 500 м/с), что значительно меньше единицы, и влиянием этого эффекта на функцию распределения по временам пребывания Е(t) можно пренебречь.
Диффузия в радиальном направлении является эффектом положительным. Она способствует выравниванию радиальных распределений параметров потока смеси и приближает характеристики реального реактора к реактору идеального вытеснения.
Третьей причиной является существование радиальной неоднородности скорости потока, обусловленной режимом течения реагирующей смеси в пространстве, ограниченным стенками: вдали от стенок скорость потока велика, а в пристеночных слоях из-за трения скорость потока падает до нуля. Радиальный профиль скорости турбулентного потока в цилиндрическом канале хорошо моделируется выражением:
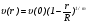 , (33)
, (33)
где R – радиус канала реактора; m = 5 для шероховатых стенок, m = 7 для гладких стенок при значениях критерия Рейнольдса 2300 Re 105 и m = 8 при Re > 105.
Если рассчитать время пребывания (0) газа в реакторе, пользуясь осевым значением скорости, то есть 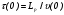 , то среднее время пребывания газа в реакторе составит [1]:
, то среднее время пребывания газа в реакторе составит [1]:
m
5
7
8
τ
1.33 τ(0)
1.22 τ(0)
1.19 τ(0)
Для ламинарного режима течения = 2(0), следовательно, необходимо отдать предпочтение сильно турбулентному режиму течения. Эти данные позволяют уточнить объем реактора, рассчитанного на заданное время пребывания в нем реагирующей смеси.
3.2 Перемешивание реагентов с энергоносителем в смесителе
Одной из особенностей ПХ-процессов является то, что часть химических превращений происходит в процессе перемешивания сырья с плазмой. Наличие этой особенности и определяет необходимость раздельного анализа и моделирования реактора и смесителя.
Смешение реагентов с энергоносителем – один из важнейших этапов ПХ-процесса. На этом этапе необходимо обеспечить прогрев сырья до требуемой температуры и перемешать реагенты настолько, чтобы каждая молекула имела возможность вступить во взаимодействие (то есть до молекулярного уровня). При этом требования к интенсивности перемешивания оказываются очень жесткими, поскольку продолжительность смешивания должна быть меньше продолжительности химической реакции ( 10-4 – 10-3с). Считается, что продолжительность перемешивания должна быть на порядок меньше продолжительности ПХ-процесса.
3.2.1 Стадии перемешивания
Используемые в ПХ-процессах реагенты могут находиться в различных агрегатных состояниях: газообразном, жидком, в виде суспензий, эмульсий, паст и твердых веществ. Часто процесс перемешивания разделяют на две стадии [1].
1-й стадией процесса перемешивания обычно является ввод и равномерное распределение отдельных частиц сырья (турбулентных глобул газа, капель жидкости, частиц порошка) по всему сечению наиболее высокотемпературной зоны энергоносителя.
Затем, на 2-й стадии, происходит перемешивание до молекулярного уровня (дробление и диффузия турбулентных глобул газа, испарение конденсированных частиц и взаимная диффузия образовавшихся паров). Следует отметить, что разделение на такие стадии является довольно условным.
На 1-й стадии, учитывая тот факт, что продолжительность испарения конденсированных частиц и диффузии глобул газа существенно возрастают с увеличением их размера, необходимо организовать процесс так, чтобы размер вводимых диспергированных частиц был невелик.
Поэтому в большинстве случаев целесообразно использовать мелкие фракции порошка сырья, применять форсунки с интенсивным дроблением жидкости, а газообразные реагенты вводить через отверстия малого диаметра. При этом желательно, чтобы распределение частиц по размерам было бы близко к монодисперсному.
На 1-й стадии компоненты сырья распределяются по объему равномерно в виде глобул, внутри которых, однако, распределение концентрации неравномерное. На 2-й стадии за счет процессов диффузии происходит выравнивание концентраций на молекулярном уровне.
При перемешивании глобулы дробятся, пока не достигнут размеров, определяемых соотношением Колмогорова:
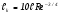 ,
,
где – характерный размер потока (диаметр трубки).
Исследования показали, что продолжительность дробления глобул до размеров 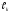 превышает время молекулярной диффузии в маштабе . Поэтому скорость процесса перемешивания определяется скоростью уменьшения размеров только крупных глобул, размер которых соизмерим с характерным размером отверстия, через которое вводится сырье.
превышает время молекулярной диффузии в маштабе . Поэтому скорость процесса перемешивания определяется скоростью уменьшения размеров только крупных глобул, размер которых соизмерим с характерным размером отверстия, через которое вводится сырье.
3.2.2 Модель перемешивания газовых потоков в цилиндрических каналах
Процессы взаимодействия турбулентных потоков с сырьем плохо поддаются теоретическому описанию (до сих пор отсутствует достаточно разработанная теория турбулентности). Поэтому основные закономерности базируются на экспериментальных результатах.
На основе анализа результатов экспериментальных и теоретических работ была предложена следующая приближенная модель процесса перемешивания газовых потоков в цилиндрических каналах (рис. 10) [1].
d
D
l0
Ln
а)
Ln max
б)
в)
Ln
Рис. 10. Схема перемешивания турбулентной струи с потоком газа в канале
Пусть в цилиндрическом канале диаметром D распространяется поток газа, в который вдувается газ из отверстия диаметром d < D, расположенного на оси потока (рис. 10а).
Предположим, что степень турбулентности в струе вдуваемого газа достаточно высока для того, чтобы распространение этой струи можно было описать с помощью полуэмпирических соотношений, предложенных для описания свободных затопленных турбулентных струй [21].
Вдуваемую струю можно считать свободной до тех пор, пока ее пограничный слой не соприкасается со стенками канала. Справедливость этого подтверждается результатами экспериментов в пределах изменения отношения D/d от 1.4 до 13.
Очевидно, что профиль концентрации вдуваемого газа в канале можно считать установившимся (в грубом масштабе), когда внешние границы вдуваемой струи пересекутся со стенками канала, при условии, что расстояние Ln, на котором происходит заполнение сечения канала пограничным слоем вдуваемой струи, не меньше длины начального участка этой струи l0.
Оценим Ln исходя из представлений полуэмпирической теории свободных турбулентных струй. Согласно этим представлениям, тангенс угла наклона внешней границы свободной турбулентной струи к ее оси составляет: tg /2 = 0.22 0.30 в широком диапазоне изменения значений числа Рейнольдса Re, скоростей и температуры турбулентной струи.
Следовательно, длина зоны установления профиля концентрации вводимого в поток газа составит величину:
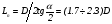 . (34)
. (34)
В крайнем случае, когда вводимая струя смещена с оси канала и располагается у ее стенки (рис. 10б), величина Ln лежит в интервале (3.4 4.6)D.
В случае поперечного ввода струи газа в поток (рис. 10в) добиться строго симметричного заполнения сечения реактора пограничным слоем холодного газа практически не удается.
Поэтому, для того чтобы обеспечить достижение минимального значения Ln в условиях радиального ввода струи, необходимо пользоваться исследованиями траекторий струй в сносящих потоках.
Например, траектории струй холодных газов в сносящем плазменном потоке аргона в канале описывается с погрешностью 12% следующим выражением:
y/d = q0.48 (x/d)0.65 , (35)
где y, x – координаты точки траектории (оси струи), отсчитываемые перпендикулярно и параллельно оси плазменного потока, мм; q = 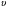 2/гг2 – отношение динамического напора струй холодного газа к среднему по сечению динамическому напору плазменной струи; , г, , г – плотности и скорости холодного и нагретого газов соответственно; d – диаметр отверстия для ввода холодного газа, мм.
2/гг2 – отношение динамического напора струй холодного газа к среднему по сечению динамическому напору плазменной струи; , г, , г – плотности и скорости холодного и нагретого газов соответственно; d – диаметр отверстия для ввода холодного газа, мм.
Здесь в качестве холодных газов использовались аргон, азот, кислород и метан, а d имел значения 0.4 1.3 мм. Существует ряд других исследований и эмпирических формул для траекторий сносимых струй.
Таким образом, согласно вышеописанной приближенной модели, длина Ln сохраняется постоянной для данного канала диаметром D и равна 2D с погрешностью 15%. Очевидно, что для сокращения длины смесителя следует уменьшить диаметр канала. Однако сокращение Ln путем уменьшения D целесообразно проводить до тех пор, пока Ln не станет сравнимой с длиной начального участка турбулентной струи  .
.
Согласно экспериментальным измерениям и теоретическим оценкам, выполненным с учетом того, что при вдуве струи в поток газа меньшей плотности несколько увеличивается, имеем = (5 7)d, где d – диаметр отверстия для вдува. Следовательно, минимальное значение Dmin составит величину, равную (2.5 3.5)d, что не противоречит одному из основных положений модели – о свободном характере распространения струи холодного газа в канале реактора.
Время установления профиля концентрации введенного в реактор газа составит величину 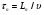 , где – линейная скорость движения смеси в канале.
, где – линейная скорость движения смеси в канале.
3.3 Закалка продуктов плазмохимических процессов.
Способы закалки
Чтобы сохранить целевые продукты, выходящие из ПХ-реактора, необходимо предотвратить обратные, параллельные или последовательные реакции, ведущие к снижению концентрации целевых продуктов. Чаще всего данную задачу решают с помощью закалки – быстрого снижения температуры системы до значений, при которых такие реакции невозможны или проходят достаточно медленно.
Закалку необходимо проводить в следующих случаях.
1) Целевой продукт является промежуточным на пути системы к равновесию, например, СН4 С2Н2 С + Н2. Закалка предотвращает наступление равновесия и останавливает развитие процесса на определенной стадии, когда достигается максимальный выход целевых компонентов. В вышеприведенном примере ацетилен (С2Н2) – это достаточно устойчивое (метастабильное) вещество при комнатной температуре.
Отметим, что в ПХ-процессе получения ацетилена достаточно опоздать с закалкой на 0.002 с, чтобы концентрация его уменьшилась в смеси с 16 до 10%.
2) Целевые продукты устойчивы (равновесны) при условиях их образования в ПХ-реакторе, то есть при высоких температурах. В области же низких (комнатных) температур равновесие смещается в сторону образования других компонентов. Концентрация целевого продукта при этом существенно снижается, а иногда близка к нулю. Закалка в данном случае «замораживает» равновесный состав, отвечающий высокой температуре.
3) Целевые продукты образуются в процессе закалки из молекул, атомов и радикалов, полученных в реакционном канале ПХ-реактора.
Требуемые скорости закалки dT/dt определяются кинетикой протекания соответствующего процесса, его механизмом, значениями кинетических параметров отдельных стадий, а также начальной температурой продуктов. Необходимое значение dT/dt может быть определено на основании кинетических расчетов или из эксперимента.
Часто при организации закалки необходимо обеспечить не просто среднюю скорость закалки в некотором диапазоне температур, но и выдержать определенную закономерность снижения температуры (например, вначале большая скорость, а в конце – малая). Скорости закалки, как правило, лежат в интервале 105 – 108 К/c.
Теплообмен при закалке имеет ряд особенностей, связанных с высокой скоростью изменения температуры:
– процесс теплообмена протекает быстро, и длительность его может оказаться соизмеримой с длительностью релаксации некоторых процессов (то есть может появиться неравновесность в распределениях по энергетическим уровням);
– изменения температуры сопровождаются химическими реакциями, и выделяемая в ходе реакций энергия может изменить температуру газа и тем самым уменьшить скорость закалки. В некоторых случаях тепловой эффект этих экзотермических реакций может настолько скомпенсировать теплоотвод в закалочном устройстве, что dT/dt может оказаться близким к нулю. Поэтому в данном случае (при значительных концентрациях атомов и радикалов) начало закалки должно предшествовать моменту достижения максимального выхода целевого продукта или же необходимо повысить интенсивность охлаждения.
В плазмохимии применяют следующие виды закалки:
1) закалка при взаимодействии продуктов реакции с твердой фазой;
2) закалка при взаимодействии продуктов реакции с жидкой фазой;
3) закалка при взаимодействии продуктов реакции с газовой фазой;
4) газодинамическая (термодинамическая) закалка.
1
2
Рис. 11. Поверхностный теплообменник: 1 − вода; 2 − трубы
Закалка первыми двумя способами осуществляется при взаимодействии продуктов как со сплошной, так и с дисперсной фазой.
Устройства для закалки твердой фазой
К таким устройствам, прежде всего, следует отнести поверхностные теплообменники. Закалку с помощью теплообменников иногда называют «поверхностной», поскольку охлаждение продуктов происходит на холодной поверхности. Поверхностная закалка применяется чаще всего. Ее можно сочетать с утилизацией тепла (например, получение пара, предварительный подогрев исходных реагентов). Котлы-утилизаторы, используемые для производства пара, могут служить закалочными устройствами, если обеспечивают необходимую скорость закалки.
Соответствующие закалочные устройства состоят из вертикально или наклонно расположенных труб (рис. 11), выполненных из металла с большой теплопроводностью и имеющих круглые, эллиптические или прямоугольные сечения. Между трубами продувается охлаждаемый газ.
Газ
Рис. 12. Закалочное устройство с перфорированными дисками
Вода
При необходимости трубы могут быть расположены в несколько рядов. При шахматном расположении труб наблюдается интенсивная турбулизация потока газа, что обусловливает более эффективную теплоотдачу и охлаждение газа.
Перспективной конструкцией считается конструкция, основным узлом которой являются перфорированные медные водоохлаждаемые диски (рис. 12). В данной конструкции достигнута скорость закалки dT/dt 2·107 К/c (в каждом из 6 дисков диаметром 4 см и толщиной 1 см находится 33 отверстия диаметром 3.6 мм).
Газ на закалку
Частицы
Рис. 13. Схема прямоточного закалочного устройства
Высокая скорость закалки достигается также при непосредственном взаимодействии газового потока с дисперсной твердой фазой (с частицами микронных и субмикронных размеров). При этом возникает возможность создавать развитую поверхность нагревания в виде непрерывно движущихся частиц, а также утилизировать тепловую энергию. Такое устройство должно обеспечивать малое время пребывания частиц в зоне высоких температур во избежание их спекания. С этой точки зрения классический «кипящий слой» мало пригоден.
Более эффективным является теплообменник с проточными дисперсными теплоносителями, которые могут работать по принципам прямотока и противотока. При прямотоке (рис. 13) увеличивается длина теплообменника, а при противотоке – его диаметр. Кроме того, при прямотоке во избежание чрезмерного уноса диаметр частиц должен быть больше, чем при противотоке.
Важным преимуществом подобных теплообменников является возможность утилизировать тепловую энергию. В этом случае частицы выступают в роли промежуточного циркулирующего теплоносителя. Он циркулирует между идущим на закалку газом (верхняя камера) и вторичным (нагреваемым) теплоносителем. Теплообменник (рис. 14) состоит из двух теплообменных камер и связывающей их транспортной системы.
Недостатками таких теплообменников являются: необходимость в устройствах ввода и вывода дисперсной среды и ее транспортировка; отделение твердых частиц от газового потока; жесткие требования к частицам по термостойкости, механической прочности, химической стойкости и др.
2
1
5
5
4
4
3
Рис. 14. Теплообмен ник с противотоком: 1 – бункер; 2 – транс-портировка частиц; 3 – распределительный узел; 4 – ввод газа; 5 – вывод газа
Устройства для закалки жидкостью
В таких устройствах процесс может быть организован двояко: сплошной фазой является газ, а дисперсной – жидкость, и наоборот.
По первому способу жидкость впрыскивается в газовый поток через форсунки. При закалке диспергированной жидкостью теплоотвод осуществляется за счет нагрева и за счет испарения капель. Выбор используемой жидкости определяется конкретным технологическим процессом.
Для достижения высоких скоростей закалки, испарения и смешения должен обеспечиваться тонкий и равномерный распыл жидкости. Для выполнения этого условия в установке размещают максимальное количество форсунок. Струи от форсунок могут распространяться радиально или аксиально каналу закалочного устройства (рис. 15).
Размещение, показанное на рис. 15б, оказывается более эффективным, так как капли жидкости более равномерно заполняют сечение закалочного канала. В схеме рис. 15а капли от радиальных форсунок оттесняются потоком от центра к стенке.
Другой вид закалки, где диспергируется охлаждаемый газ, осуществляется в устройствах барботажного типа (рис. 16), скорость закалки в которых достигает 105–106 К/с. Удельная поверхность теплопередачи (поверхность газовых пузырьков), а, следовательно, и скорость закалки увеличиваются с уменьшением диаметра пузырьков.
Вода
Рис. 15. Схема размещения форсунок в закалочном устройстве:
а) радиальное; б) аксиальное
а)
б)
Поэтому газовый поток дробят при помощи распределительных решеток.
При правильном подборе закалочной жидкости получают дополнительный выход целевых продуктов за счет утилизации тепловой энергии газового потока. Так, при закалке продуктов пиролиза природного газа жидкими углеводородами был получен дополнительный выход ацетилена.
Холодный газ
D
d
Газ на закалку
Рис. 17. Закалка холодными струями
Газ
Вода
Парогазовая смесь
Рис. 16. Закалочное устройство барботажного типа
Скорость закалки продуктов струями холодного газа (рис. 17) лимитируется интенсивностью перемешивания горячего газа и охлаждающего агента, при этом скорость охлаждения достигает 106–107 K/c. Холодный газ вводится обычно струями, направленными под некоторым углом к охлаждающему потоку (обычно радиально или тангенциально).
В данном закалочном устройстве рекомендуемое соотношение d/D 0.3 0.4. В правильно рассчитанной конструкции смешение заканчивается на длине (12)D. Недостатком данных конструкций является разбавление продуктов реакции охлаждающим газом. Поэтому, по возможности, для охлаждения используется тот же пиролизный газ, прошедший стадию охлаждения в котлах-утилизаторах. При этом наряду с устранением разбавления продуктов происходит утилизация тепла.
Газодинамическая закалка.
При адиабатическом течении газа (то есть без теплообмена с окружающей средой) полная энергия какой-либо массы газа остается неизменной во времени:
Нп + М2/2 = Const,
где Нп – полная энтальпия массы газа М.
Газ на закалку
Свехзвуковое сопло (Лаваля)
Газовая турбина
Охлажденый газ
Генератор тока
Рис. 18. Газодинамическое закалочное устройство
Реакционные газы поступают на закалку в сверхзвуковое сопло (Лаваля), где охлаждаются до заданной температуры за счет превращения тепловой энергии в кинетическую при ускорении газового потока. Далее газ поступает на лопасти газовой турбины, где кинетическая энергия газа преобразуется в электрическую энергию, выработанную генератором, соединенным с турбиной.
Для газодинамической закалки необходим перепад давлений в 23 МПа. Кроме того, при торможении газового потока его температура вновь возрастает. Поэтому такой способ закалки можно применять только в комбинации с другими способами.
Рассмотренные способы закалки достигают следующих скоростей:
1) Поверхностный теплообменник
105 107 K/c.
2) Твердыми частицами
106 107 K/c.
3) Распыленная жидкость
105 107 K/c.
4) Барботажный
104 105 K/c.
5) Смешением газов
105 106 K/c.
6) Газодинамический
106 108 K/c.
Список литературы
Теоретическая и прикладная плазмохимия / Л.С. Полак, А.А. Овсянников, Д.И. Словецкий и др. М.: Наука, 1975. 304 с.
Методы исследования плазмы / Под ред. В. Лохте-Хольтгревена. М.: Мир, 1971. 552 с.
Физика и техника низкотемпературной плазмы / Под ред. С.В. Дресвина. М.: Атомиздат, 1972. 352 с.
Жуков М. Ф. Прикладная динамика термической плазмы / М.Ф. Жуков, А.С. Коротеев, Б. А. Урюков. Новосибирск: Наука, 1975. 300 с.
Жуков М.Ф. Электродуговые нагреватели газа (плазмотроны) / М. Ф. Жуков, В.Я. Смоляков, Б.А. Урюков. Новосибирск: Наука, 1973. 232 с.
Плазмохимическая переработка угля / М.Ф. Жуков, Р.А. Калиненко, А. А. Левицкий, и др. М: Наука, 1990. 200 с.
Сурис А.Л. Плазмохимические процессы и аппараты / А. Л. Сурис. М.: Химия, 1989. 304 с.
Бугаенко Л. Т. Химия высоких энергий / Л. Т. Бугаенко, М. Г. Кузьмин, Л.С. Полак. М.: Химия, 1988. 368 с.
Оулет Р. Технологическое применение низкотемпературной плазмы / Р. Оулет, М. Барбье, П. Черемисинофф. М.: Энергоатомиздат, 1983. 144 с.
Туманов Ю. Н. Современное состояние развития плазменных процессов в технике, технологии и металлургии / Ю.Н. Туманов // Химия плазмы. Вып. 13 / Под ред. Б. М. Смирнова. М.: Энергоатомиздат, 1986. С. 163.
Плазменная технология в производстве СБИС: Пер. с англ. / Под ред. Н. Айнспруга и Д. Брауна. М.: Мир, 1987. 469 с.
Бородин В.И. Установка для обработки воды плазмой тлеющего разряда /В. И. Бородин // Материалы конф. по физике низкотемпературной плазмы ФНТП-2001. Петрозаводск, 2001. С. 197–201.
Туманов Ю.Н. Низкотемпературная плазма и высокочастотные электромагнитные поля в процессах получения материалов для ядерной энергетики / Ю.Н. Туманов. М.: Энергоатомиздат, 1989. 280 с.
Низкотемпературная плазма. Т. 8: Плазменная металлургия. Новосибирск: Наука, 1992. 265 с.
Низкотемпературная плазма. Т. 3: Химия плазмы. Новосибирск: Наука, 1991. 428 с.
Низкотемпературная плазма. Т. 4: Плазмохимические технологии. Новосибирск: Наука, 1991. 454 с.
Ragnar Bernhard, Jarl Martensson Plasmaforgasning av tjocklut ersatter sodapanna och kalkcykel / Ragnar Bernhard, Jarl Martensson // Svensk papperstidning. 1985. № 3. P. 26–27.
Бородин В.И. Термодинамический анализ высокотемпературных процессов переработки материалов: Методич. указ. к лабораторным работам / В.И. Бородин, В.А. Трухачева. Петрозаводск, 1998. 40 с.
Бородин В.И. Физика и техника дугового разряда высокого давления: Учеб. пособие / В.И. Бородин. Петрозаводск, 1999. 104 с.
Зельдович Я.Б. Физика ударных волн и высокотемпературных гидродинамических явлений / Я.Б. Зельдович, Ю.П. Райзер. М.: Наука, 1966. 582 с.
Абрамович Г.Н. Прикладная газовая динамика / Г.Н. Абрамович. М.: Наука, 1969. 824 с.

Нравится материал? Поддержи автора!
Ещё документы из категории физика:
Чтобы скачать документ, порекомендуйте, пожалуйста, его своим друзьям в любой соц. сети.
После чего кнопка «СКАЧАТЬ» станет доступной!
Кнопочки находятся чуть ниже. Спасибо!
Кнопки:
Скачать документ