Синдром Клайнфельтера
 МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ УКРАИНЫ
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ УКРАИНЫ
ХАРЬКОВСКИЙ НАЦИОНАЛЬНЫЙ УНИВЕРСИТЕТ
ИМ. В.Н. КАРАЗИНА
Биологический факультет
кафедра генетики и цитологии
Курсовая работа
Синдром Клайнфельтера
Выполнила
студентка IVкурса
биологического факультета
группы БГ-41
Бабенко Е.С,
Харьков 2010г.
Синдром Клайнфельтера включает в себя целую группу патологий. Данное заболевание было описано одноименным ученым в 1942 году. Описание заключалось в том, что Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички.
К сожалению, литературные источники, использованные мною, расходятся во мнении, когда и кем была доказана цитогенетическая природа возникновения синдрома Клайнфельтера. Так же существует множество вариантов частоты встречаемости заболевания, что я подробнее опишу далее.
Пожалуй, единственное в чем не возникло существенных разногласий у авторов книг и статей – это то, что люди, страдающие данной патологией могут вести нормальный образ жизни, ничем не отличаться от остальных в случае своевременной диагностики и назначения лечения.
В связи с ухудшением экологической обстановки, количество детей рождаемых с различными патологиями возрастает. При Синдроме Клайнфельтера диагностика затруднена, это связано с тем, что ребенок рождается с виду нормальным, симптомы заболевания начинают проявляться, как правило, только в препубертатном периоде, когда лечение уже не настолько эффективно. Диагностика заболевания до препубертатного периода возможна только цитогенетическими методами. Как известно, в нашей стране нет возможности кариотипирования всех новорожденных детей и как правило к генетическому анализу кариотипа прибегают только тогда, когда уже проявляются видимые симптомы.
В связи с выше сказанным, я считаю изучение данной патологии и др. заболеваний связанных с затрудненной диагностикой в раннем возрасте очень актуальным.
История открытия
Как уже отмечалось выше, Синдром Клайнфельтера был описан в 1942 году, на основе схожей симптоматики группы пациентов (первичный мужской гипогонадизм), было предложено лечение гормонами, но цитогенетические основы заболевания оставались неизвестны.
По одним данным, цитогенетическая основа синдрома Клайнфельтера впервые в 1956 году была описана Бриге и Баром. В кариотипе больных они выявили лишнюю Х-хромосому (полисомия по Х-хромосоме), таким образом, их кариотип представлял собой 47 XXY [2].
По другим сведения цитогенетическая основа синдрома Клайнфельтера была доказана в 1959 году Джакобсом и Стронгом. Ими было установлено наличие дополнительной Х-хромосомы при синдроме Клайнфельтера [3].
Активные исследования данной патологии велись в 70-х годах в США. Всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить частоту встречаемости и генетические особенности синдрома. Было установлено, что далеко не всегда синдром Клайнфельтера вызван нарушениями количества хромосом.
Не смотря на различия в хронологии, открытия были сделаны и цитогенетические основы заболевания были установлены, что дало возможность осуществлять диагностику на основании методики исследования кариотипа.
Диагностика синдрома Клайнфельтера
Диагностика синдрома Клайнфельтера затруднена, в связи с тем, что в основном симптоматика проявляется в препубертатном периоде, а в случае мозаичных форм (чаще всего с кариотипом 46,XY/47,XXY) симптомы могут не проявляться вообще. Больные мозаичными формами в некоторых случаях могут иметь потомство.
Диагностика любой хромосомной патологии может осуществляться несколькими способами (в нашем случае речь идет о цитогенетических методах и о диагностике по симптомам). Следует помнить, что на основе симптоматической диагностики можно только предположить синдром Клайнфельтера, а не однозначно его диагностировать.
Пренатальная диагностика осуществляется методом амниоцетеза или боипсии хориона.
Амниоцентез - это одно из важнейших достижений пренатальной диагностики. С его помощи получают околоплодную жидкость для исследования. Клетки плода, химические соединения и микроорганизмы в жидкости, окружающей плод, несут большой объем информации о новом человеческом существе. При помощи анализа можно выяснить генетическую структуру плода, состояние его здоровья, а также степень развития.
Проведение этой процедуры рекомендуется, если:
мать в возрасте старше 35 лет (амниопункция проводится обычно с целью определения синдрома Дауна);
семья уже имеет ребенка с синдромом Дауна или с синдромом Хантера. Пара имеет ребенка или близкого родственника с нарушениями нервной системы;
мать является носителем гемофилии, которую она может передать только сыновьям (при помощи амниопункций можно определить пол ребенка, но не факт наследования гена, несущего гемофилию);
оба родителя страдают болезнью Тэй-Сакса или серпосерповидно-клеточной анемией;
требуется определение степени развития легких ребенка, так как они начинают самостоятельно функционировать последними;
известно, что один из родителей болен хореей Гентингтона;
исследование околоплодной жидкости в этом случае необходимо для определения нарушений плода.
Диагностическая амниопункция беременности производится обычно в срок беременности между 16и неделями, иногда даже уже на 14-й, а бывает - и на 20-й неделе. Но ученые разрабатывают возможности проведения амниопункции в более ранние сроки - между 10 и 14 неделями.
Амниопункцию можно также производить в последнем периоде беременности с целью определения степени развития легких у плода.
Биопсия хориона
Как метод пренатальной диагностики биопсия хориона может определить врожденные дефекты плода на очень ранних сроках беременности, когда аборт может быть менее сложным, поэтому она обретает все большую популярность. Хорион - это плодовая оболочка.
Биопсия хориона может применяться экспериментально вместо амниопункции. Бывают такие случаи, когда из-за малого количества околоплодной жидкости невозможна амниопункция. К тому же биопсия хориона позволяет значительно быстрее получить результат, и в будущем она сделает возможным лечение и коррекцию многих нарушений развития плода еще в матке.
Биопсия хориона применяется для выявления только таких нарушений, как синдром Тэй-Сакса, синдром Клайнфельтера, серповидно-клеточная анемия, большинство видов муковисцидоза и синдром Дауна в тех случаях, когда эта болезнь уже проявлялась прежде в семье или известно, что родители являются носителями этой болезни.
Это исследование проводится по тем же указаниям, что и в случае амниопункции, только для оценки развития легких у плода биопсия хориона не применяется. Влагалищная биопсия хориона проводится между 8 и 12 неделями беременности, а также между 9 и 11 неделями при брюшинной биопсии. Брюшинную биопсию можно применять в исключительных случаях во втором и третьем триместре беременности.
Это исследование проводится только в центральных медицинских учреждениях через влагалище, шейку матки и брюшную стенку. Эти процедуры болезненны. Они могут вызвать легкое или серьезное недомогание.
Методы пренатальной диагностики при направильном проведении манипуляции могут нанести существенный вред плоду [4].
Постнатальная диагностика
Определение Х- и Y-хроматина часто называют методом экспресс диагностики пола. Исследуют клетки слизистой оболочки ротовой полости вагинального эпителия или волосяной луковицы. В ядрах клеток женщин в диплоидном наборе присутствуют две хромосомы X, одна из которых полностью инактивирована (спирализована, плотно упакована) уже на ранних этапах эмбрионального развития и видна в виде глыбки гетерохроматина, прикреплённого к оболочке ядра. Инактивированная хромосома X называется половым хроматином или тельцем Барра. Для выявления полового Х-хроматина (тельца Барра) в ядрах клеток мазки окрашивают ацетарсеином и препараты просматривают с помощью обычного светового микроскопа. В норме у женщин обнаруживают одну глыбку Х-хроматина, а у мужчин её нет.
Для выявления мужского Y-полового хроматина (F-тельце) мазки окрашивают акрихином и просматривают с помощью люминисцентного микроскопа. Y-хроматин выявляют в виде сильно светящейся точки, по величине и интенсивности свечения отличающейся от остальных хромоцентров. Он обнаруживается в ядрах клеток мужского организма.
Отсутствие тельца Барра у женщин свидетельствует о хромосомном заболевании - синдроме Шерешевского-Тернера (кариотип 45, ХО). Присутствие у мужчин тельца Барра свидетельствует о синдроме Клайнфельтера (кариотип 47, XXY).
Определение Х- и Y-хроматина - скрининговый метод, окончательный диагноз хромосомной болезни ставят только после исследования кариотипа.
Кариотипирование
Для изучения хромосом чаще всего используют препараты кратковременной культуры крови, а также клетки костного мозга и культуры фибробластов. Доставленную в лабораторию кровь с антикоагулянтом подвергают центрифугированию для осаждения эритроцитов, а лейкоциты инкубируют в культуральной среде 2-3 дня. К образцу крови добавляют фитогемагглютинин, так как он ускоряет агглютинацию эритроцитов и стимулирует деление лимфоцитов. Наиболее подходящая фаза для исследования хромосом - метафаза митоза, поэтому для остановки деления лимфоцитов на этой стадии используют колхицин. Добавление этого препарата к культуре приводит к увеличению доли клеток, находящихся в метафазе, то есть в той стадии клеточного цикла, когда хромосомы видны лучше всего. Каждая хромосома реплицируется (производит свою копию) и после соответствующей окраски видна в виде двух хроматид, прикреплённых к центромере, или центральной перетяжке. Затем клетки обрабатывают гипотоническим раствором хлорида натрия, фиксируют и окрашивают.
Для окраски хромосом чаще используют краситель Романовского-Гимзы, 2% ацеткармин или 2% ацетарсеин. Они окрашивают хромосомы целиком, равномерно (рутинный метод) и могут быть использованы для выявления численных аномалий хромосом человека.
Для получения детальной картины структуры хромосом, идентификации (определения) отдельных хромосом или их сегментов используют различные способы дифференциального окрашивания. Наиболее часто применяют методы Гимза, а также G- и Q-бендинга. При микроскопии препарата по длине хромосомы выявляется ряд окрашенных (гетерохроматин) и неокрашенных (эухроматин) полос. Характер поперечной исчерченности, получаемый при этом, позволяет идентифицировать каждую хромосому в наборе, так как чередование полос и их размеры строго индивидуальны и постоянны для каждой пары.
Метафазные пластинки отдельных клеток фотографируют. Из фотографий вырезают индивидуальные хромосомы и наклеивают их по порядку на лист бумаги; такая картина хромосом называется кариотипом.
Применение дополнительного окрашивания, а также новые методы получения хромосомных препаратов, позволяющих растягивать хромосомы в длину, значительно увеличивают точность цитогенетической диагностики.
Для описания кариотипа человека разработана специальная номенклатура. Нормальный кариотип мужчины и женщины обозначают как 46, XY и 46, XX соответственно. У больных синдромом Клайнфельтера в кариотипе присутствует добавочная Х или У хромосома (в некоторых случаях добавочных хромосом может быть несколько) [6].
Клинические проявления
Синдром Клайнфелтера относится к группе хромосомных болезней. Под описанные одноименным ученым симптомы подходит целая группа заболеваний, поэтому синдром Клайнфелтера разделяют на истинный (хроматинположительный) и ложный (хроматинотрицательный).
Хроматинположительный синдром Клайнфелтера характеризуется тем, что в буккальных мазках определяется половой хроматин по женскому типу. Таких пациентов иногда называют ХХУ-мужчинами, для более точного описания синдрома. Как уже отмечалось, заболевание обусловлено хромосомной аномалией. У больных чаще всего есть одна лишняя X-хромосома, реже — несколько Х-хромосом (кариотип 47XXY; 48XXXY; 49XXXXY). Интеллект больных синдромом Клайнфелтера часто снижен, предполагают, что степень его нарушения пропорциональна числу добавочных Х-хромосом в кариотипе.
Встречается также хроматинотрицательная форма синдрома Клайнфелтера с кариотипом 46 XY, характеризующаяся постпубертатной атрофией яичек и врожденным отсутствием герминативных клеток. Описан еще синдром Клайфелтера с кариотипом 47 XYY, при котором дисгенезия семенных канальцев наблюдается весьма редко; распознавание его возможно на основе определения мужского полового хроматина по Y-тесту флюоресцентным методом (Gasperson и соавт., 1969; Lech, 1969). При этом обнаруживаются два флюоресцирующих тельца мужского полового хроматина. Существуют две разновидности хроматинотрицательной формы синдрома Клайнфелтера. Первая характеризуется постпубертатной атрофией яичек. Отмечаются гиалиноз канальцев, лишенных семяродного эпителия, обилие в них эластической ткани, канальцевый фиброз, скопление интерстициальных эндокриноцитов. Для второй разновидности типично отсутствие герминативных элементов, хотя в канальцах яичка содержатся поддерживающие (сертолиевы) клетки. Гиалиноз канальцев встречается редко, периканальцевый фиброз не развивается.
В свою очередь в истинном (хроматинположительном) синдроме по клиническому проявлению выделяют две основные формы: эндоморфную и экзоморфную. Эндоморфная форма характеризуется нормальным развитием половых органов и вторичных половых признаков, наличием гинекомастии и некоторым отставанием роста. При экзоморфной форме отмечаются евнухоидное телосложение, недоразвитие половых органов (половой член часто бывает малых размеров, яички плотные и маленькие) и вторичных половых признаков [8].
В ряде случаев выявляется полисомия по Y-хромосоме при моносомии по Х-хромосоме, а также полисомия по Х- и Y-хромосоме (Г. Г. Мирзаянц). У больных с мозаицизмом половые хромосомы могут быть разными в различных тканях. В связи с этим заболевание может выявляться и при отрицательном половом хроматине. Наиболее часто мозаицизм представлен 46XY/47XXY. В эмбриональный период формирование яичек и мужских половых органов происходит нормально, что обусловлено наличием в кариотипе Y-хромосомы.
Клиническая картина синдрома смазана, однако существует целый ряд симптомов характеризующих данную патологию. Для начала необходимо привести статистические данные о частоте встречаемости.
Статистические данные неоднозначны, частота встречаемости отмечается как 1 случай на 500-1000 живорожденных мальчиков. Отмечается, что частота заболеваемости у мужчин с нормальным фенотипом составляет 1:1100 мужчин, среди больных шизофренией – 1:300, олигофренией 1:95 случаев. Т.к. синдром Клайнфельтера практически в 100% случаев сопровождается бесплодием, по статистике у 1 из 10 мужчин страдающих бесплодием обнаруживается синдром Клайнфельтера [7].
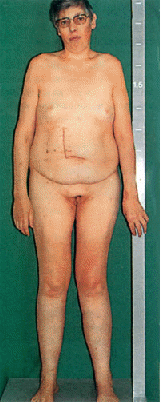
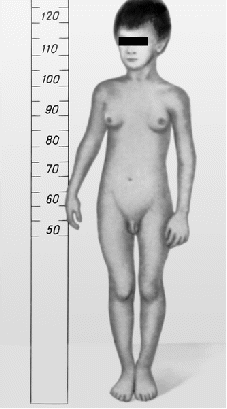
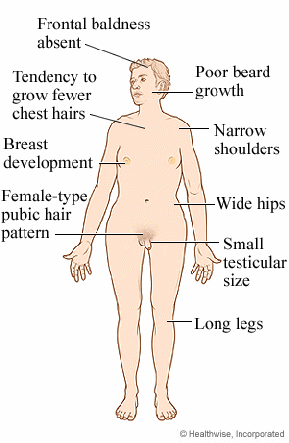
Видимые отклонения, возникающие при синдроме Клайнфельтера, я рассмотрю на примере иллюстраций (рис.1 - рис.3).
Рис. 1
Рис. 2
Рис. 3
На первой иллюстрации продемонстрирован стандартный вид больного. Можно видеть, что типичным для данной патологии является неполная маскулинизация (т.е. развитие внешних признаков идет по женскому типу). Больные характеризуются высоким ростом (как правило выше 1,95м), телосложение евнухоидное (узкие плечи, широкие бедра), молочные железы также сформированы по женскому типу, зачастую увеличены (гинекомастия), оволосение на теле слабо или отсутствует. Пожалуй, самым важным симптомом является уменьшение гонад, при этом мошонка и половой член нормального размера.
На иллюстрации 2 и 3 представлены больные, которые по-видимому имеют в кариотипе 2 и более добавочные Х или У хромосомы, т.к. степень выраженности симптомов значительная (связь проявления симптомов и наличие добавочных хромосом я опишу далее). На иллюстрации 2, заметен явно выраженный гипогонадизм, но при этом можно наблюдать значительное уменьшение в размерах полового члена, оволосение отсутствует, телосложение евнухоидное (экзоморфная форма). На рисунке 3, представлен мальчик 6 лет, видно атипично раннее проявление симптомов (в допубертатном возрасте).
Гистологически обнаруживаются дегенерация герминативного эпителия и гиалиноз семенных канатиков. Больные бесплодны (азооспермия, олигоспермия).
Подводя итог, патология характеризуется:
смазанной клинической картиной;
характерен высокий рост;
гинекомастия;
слабое оволосение лица, подмышечных впадин и лобка;
бесплодие;
в мазках буккального эпителия обнаруживаются глыбки полового хроматина;
гиперплазией лейдиговских клеток с нормальной или умеренно сниженной их функцией и увеличением секреции фолликулостимулирующего гормона и др.
При синдроме Клайнфелтера нередко можно наблюдать негативные черты в поведении больных. Характерными для них являются вялость, пассивность, утрата инициативы, отсутствие выраженного интереса к окружающему, признаки безвольности, замкнутость, мнительность, склонность к аффективным вспышкам. В некоторых случаях проявляется тенденция к бродяжничеству— уход из дома, злоупотребление алкоголем, совершение антиобщественных поступков и правонарушений (кража, поджог) . Торможение преобладает над возбуждением
Так же, существует группа симптомов, которые называют сопутствующими (т.е. свойственными для данной аномалии, но не являющимися важными в процессе диагностики). К таким симптомам относятся:
бесплодие;
неполная маскулинизция;
уменьшенное либидо;
остеопороз;
тауродонтизм («Бичьи зубы»);
болезни венозной системы;
нарушение поведение;
аутоиммунные заболевания;
снижение подвижности;
низкая самооценка;
повышенная раздражительность;
высокий рост и склонность к полноте;
нарушение моторных функций и развития.
Специфика проявления симптомов может варьировать в зависимости от количества добавочных хромосом.
Цитогенетические особенности синдрома Клайнфельтера
В данном разделе я рассмотрю истинный синдром Клайнфельтера, т.к. ложный не имеет цитогенетических особенностей (кариотип больных 46 ХУ) и как правило развивается на фоне перенесенного в раннем возрасте паротического орхита [9].
Хромосомные аномалии, приводящие к синдрому Клайнфельтера, возникают в результате нарушения деления половых клеток и образования гамет, содержащих большее, чем в норме, число хромосом (явление анеуплоидии) [10].
Избыточное число половых хромосом в кариотипе приводит к дисбалансу генов. При увеличении числа лишних хромосом степень выраженности генного дисбаланса усугубляется, а клинические проявления заболевания становятся все более выраженными. Причиной болезни может стать также аномалия деления оплодотворенной яйцеклетки (зиготы), при котором наблюдается мозаичный вариант с кариотипом 46, XY/47, XXY
Не смотря на то, что иногда возникают различные вариации соединения ХХУ – хромосом, наиболее часто встречается ХУ/ХХУ – мозаицизм. При таком варианте только некоторые клетки в организме мужчины имеют дополнительную Х-хромосому, а остальные содержат нормальный набор 46ХУ. Количество клеток с лишней хромосомой отличается в каждом конкретном случае. Иногда встречаются случаи мозаицизма, при котором достаточное количество клеток имеет нормальный кариотип и поэтому такие больные способные к оплодотворению
В литературе описаны случаи, когда мужчины с синдромом Клайнфельтера имели 2 или даже 3 дополнительные хромосомы. Эти больные имели значительно более тяжелые проявления синдрома, включая низкий уровень умственного развития или различную степень отставания речи.
Наиболее редки случаи, когда у мужчин лишними оказывались не только Х, а и У – хромосомы.
Ниже приведу наиболее характерные признаки различных вариаций синдрома Клайнфельтера:
46, ХУ/47, ХХУ (Мозаицизм)
Наиболее распространенная разновидность заболевания, аномальные клоны присутствуют не во всех клетках и тканях организма, а, следовательно в буккальных мазках половой может не обнаруживаться. Проявления патологии, как правило не значительны, многие больные даже не имеют представления о наличии аномалии. Если количество аномальных клеток незначительно такие мужчины могут быть способны к оплодотворению.
47, ХХУ
Все клетки содержат добавочную Х – хромосому. Такие больные бесплодны. Данная разновидность не характеризуется задержкой умственного развития. Характерен высокий рост, евнухоидное строение тела, гипогонадотропный гипогонадизм, уменьшенное оволосение на теле и лице, гинекомастия.
48, ХХУУ
Как правило, больные имеют среднюю степень умственного отставания, высокий рост, евнухоподобное строение тела, редкие волосы на лице и теле, выраженную гинекомастию, длинные и тонкие ноги, гипогонадотропный гипогонадизм.
48, ХХХУ
Характеризуется средней степенью умственного отставания, задержка речи, замедление моторных функций, плохая координация, нарушение поведения, нормальный или высокий рост. Свойственны необычны черты лица (эпикант, гипертелоризм, выступающие губы), гипогонадизм, гинекомастия, гипоплазия полового члена, бесплодие, клинодактилия.
49, ХХХУУ
Умственное отставание колеблется от средней до тяжелой степени, характерны резкие смены поведения от пассивной до агрессивной, высокий рост, дизморфические черты лица, гинекомастия и гипогонадизм.
49, ХХХХУУ
Классическая триада со средним или тяжелым умственным отставанием, гипогонадотропным гипогонадизмом. Другие клинические проявления включаются в себя тяжелую задержку речи, проблемы с поведением, маленький вес при рождении, маленький рост, аномальные черты лица (округлое лицо в детстве, которое приобретает грубые черты с возрастом, гипертелоризм, эпикант), короткую шею, врожденные проблемы сердца, аномалии скелета, мышечную гипотонию, гиперподвижность суставов, гипоплазию половых органов и крипторхизм.
Лечение
Лечение синдрома Клайнфельтера включает сочетание лекарственных средств, гормональных препаратов и хирургического вмешательства. Корригирующая витамино- и гормонотерапия, начиная с 5—6 лет и продолженная в препубертатный и пубертатный периоды, приводит к остановке атрофии яичек и сохранению фертильности (в случае мозаичости). При нерезко выраженной гинекомастии можно рекомендовать гимнастику, в противном случае показана мастэктомия. Лечение необходимо проводить с учетом лабильности психики и повышенной мнительности этой категории больных. Больные нуждаются в пожизненной заместительной гормональной терапии.
Поэтому чрезмерное фиксирование внимания на неполноценности половой системы может оказывать психотравмирующее действие и приводить к половой несостоятельности. Последовательное и четкое проведение названных лечебных мероприятий позволяет в какой-то степени купировать как генеративную, так и копулятивную недостаточность.
Вывод
Синдром Клайнфельтера – хромосомная аномалия, характеризующаяся полисомией по половым хромосомам. Возможны различные вариации заболевания – полисомия по Х-хромосоме, полисомия по У-хромосоме при моно- и полисомии по хромосоме Х. Встречаются различные комбинации хромосом, наиболее тяжелые симптомы наблюдаются при кариотипе 49, ХХХХУУ. Наиболее распространен мозаичный кариотип 46, ХУ/ 47, ХХУ, больные могут иметь нормальный кариотип и даже не догадываться о наличии аномалии.
Клиническая картина заболевания смазана, к основным симптомам относят высокий рост, евхнухоидное телосложение, гинекомастию, слабое оволосение. С увеличением количества добавочных Х-хромосом повышается умственная отсталость.
Диагностика в основном осуществляется в препубертатном периоде, в тяжелых случаях симптомы могут проявиться на более ранних стадиях онтогенеза. Для диагностики используются цитогенетические методы.
При своевременном лечении возможно остановить атрофию яичек и содействовать нормальному формированию мужского фенотипа. Половая функция у больных синдромом Клайнфельтера, как правило, не страдает, но ведет к бесплодию (в некоторых случая больны неспособны к сперматогенезу).
Данная хромосомная аномалия не влияет на продолжительность жизни (за исключением крайне тяжелых форм, при которых у больных наблюдаются множественные пороки развития внутренних органов), следует помнить, что уязвимой стороной таких мужчин является психика. Они склонны к депрессии, суициду, но очень часто подобные расстройства сменяются неконтролируемой агрессией направленной на окружающих.
Список литературы
Михасюк О.О. Синдром Клайнфельтера, Інформація для батьків – http://downsyndrome.at.ua/pub/11
C. Schirren, J. O. Toyosi – Testicular histology in Klinfeltr’s syndrome – www.springerlink.com/content
Ворсанова С.Г. Хромосомные синдромы и аномалии. Классификация и номенклатура / С.Г. Ворсанова, Ю.Б. Юров, В.Н. Чернышев. – Ростов - на- Дону: Изд-во Ростовского ун-та, 1999.
Георгиева Д.Н. Пренатальная диагностика на ранних сроках беременности www.rosmedzdrav.ru
Энглевский Н.А. - 25.01.2010 г – Проблемы и перспективы современной медицины, биологии и экологии, Фундаментальные науки и практика Т.1 - http://tele-conf.ru/nasledstvennyie-morfologicheskie-kletochnyie-fakto/obzor-issledovaniy-polovogo-hromatina.html
Heidi Chial. Cytogenetic Methods in Diagnosing Genetic Disorders – http://www.nature.com/scitable/topicpage/cytogenetic-methods-in-diagnosing-genetic-disorders-875
Michael J. Sexton, Klinefelter Syndrome
http://www.cigna.com/
Фомина О.П. «Справочник педиатра» -
http://www.dopomoha.kiev.ua/pediatr/index.html
Г.И. Бужиевская «Основы медицинской генетики» - Киев, здоровье - 2001, 135стр
Билич Г.Л. Большой толковый медицинский словарь в 2-х томах. 2001 г – М.АСТ
Dr Jeannie Visootsak and Prof John M. Graham Klinefelter Syndrome and Its variants March 2003 http://www.orpha.net
Cammarata M, Di Simone PD, Graziano L, et al. Letter to the editor: Rare sex chromosome aneuploidies in human: report of six patients with 48,XXYY, 49,XXXXY, and 48,XXXX karyotypes. Am J Med Genet. 1999; 85: 86-87. http://www.orpha.net
Campbell WA, Price WH. Venous thromboembolic disease in Klinefelter’s syndrome. Clin Genet. 1981;19:275-280. http://www.orpha.net
Синдром Клайнфельтера http://humbio.ru/humbio/eclin/001dee20.htm
Жуковский М.А. Нарушения полового развития / М.А. Жуковский. – М.: Медицина, 1989 г.
. Синдром Клайнфельтера. http://ru.wikipedia.org/wiki/klainfeltera

Нравится материал? Поддержи автора!
Ещё документы из категории медицина, здоровье:
Чтобы скачать документ, порекомендуйте, пожалуйста, его своим друзьям в любой соц. сети.
После чего кнопка «СКАЧАТЬ» станет доступной!
Кнопочки находятся чуть ниже. Спасибо!
Кнопки:
Скачать документ