«использование нформационных технологий в молекулярной биологии»









 БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Выпускная работа по
«Основам информационных технологий»
Магистрант кафедры молекулярной биологии Бадалян Ольга Руководители: доцент Николайчик Евгений Артурович, Ph. D., ст. преподаватель Шешко Сергей Михайлович
Минск – 2010 г.
СПИСОК ОБОЗНАЧЕНИЙ КО ВСЕЙ ВЫПУСКНОЙ РАБОТЕ
ДНК
Дизоксирибонуклеиновая кислота
ИТ
Информационные технологии
ПЦР
Полимеразная цепная реакция
РНК
Рибонуклеиновая кислота
РЕФЕРАТ НА ТЕМУ «ИСПОЛЬЗОВАНИЕ НФОРМАЦИОННЫХ ТЕХНОЛОГИЙ В МОЛЕКУЛЯРНОЙ БИОЛОГИИ»
Современная молекулярная биология и молекулярная генетика определяют развитие как фундаментальной, так и прикладной биологии. Огромное разнообразие подходов к решению задач исследований требует все более точных и быстрых методов анализа. Эффективность исследований зависит от нескольких параметров, основополагающими из которых, являются техническое оснащение лаборатории и освоение новых методических подходов.
На каждом этапе работы молекулярный биолог сталкивается с применением информационных технологий. Молекулярная биология изучает такие молекулы как ДНК, РНК и белки. Такие объекты изучения нельзя потрогать руками, нельзя увидеть невооруженным глазом. Поэтому практически все методы, которые используются в молекулярной биологии, связаны с использованием информационных технологий (ИТ).
Любой молекулярно-биологический эксперимент можно разделить на три стадии: планирование эксперимента, его проведение и анализ полученных результатов. На той или иной стадии молекулярный биолог сталкивается с использованием ИТ-инструментов, будь то пакет Microsoft Office и глобальная сеть «Интернет» или же программное обеспечение для приборов, используемых в молекулярно-биологической лаборатории (амплификаторы в режиме реального времени, секвенаторы, системы гель-документации, спектрофотометры и др.). Также стоит отметить применение различных графических редакторов и программ для моделирования не только макромолекул, но и целых процессов протекающих на молекулярном уровне.
Глава 1
Анализ последовательностей ДНК
Молекулярная биология – это комплекс биологических наук, изучающих механизмы хранения, передачи и реализации генетической информации, строение и функции нерегулярных биополимеров (белков и нуклеиновых кислот). Поскольку ДНК является материальным носителем генетической информации, молекулярная биология значительно сблизилась с генетикой, и на стыке образовалась молекулярная генетика, являющаяся одновременно разделом генетики и молекулярной биологии. Для анализа генетической информации привлекается вычислительная техника, в связи с чем появились новые направления молекулярной генетики, которые иногда считают особыми дисциплинами: биоинформатика, геномика и протеомика.
Анализ последовательностей биологических макромолекул (ДНК, РНК, белков) является существенной частью повседневной работы современного биолога. Из-за большого размера макромолекул в подавляющем большинстве случаев такой анализ требует использования специализированных программ для просмотра аннотированных последовательностей, выявления интересующих исследователя функциональных участков, рестрикционного анализа, подбора праймеров для ПЦР и т.д. Кроме того, планирование молекулярно-биологических экспериментов in silico перед их реальной постановкой in vitro способно сэкономить время, средства и помочь избежать ошибок[1].
В связи с большим разнообразием возможных вариантов анализа последовательностей ДНК и потребностей молекулярных биологов в настоящее время существует значительное количество компьютерных программ, в той или иной степени эти потребности удовлетворяющих. Существующие программные решения можно разделить на две группы: крупные программные пакеты, способные выполнять всесторонний анализ последовательностей ДНК, и специализированные программы, предназначенные для решения конкретной частной проблемы. В первом случае речь идет, как правило, о наборе небольших программ (например, пакет EMBOSS [2]) суммарно представляющих мощный аналитический инструмент, который, однако, из-за слабой связи этих программ между собой и нестандартного интерфейса не очень удобен для молекулярного биолога, не специализирующегося в области биоинформатики. Программы второй группы имеют, как правило, более привычный простой графический интерфейс и должны быть удобнее в работе (при условии реализации всех необходимых для пользователя функций). Также существуют различные web-приложения, среди которых можно, наверное, найти программы для всех мыслимых вариантов анализа последовательностей биологических макромолекул, но подавляющее большинство таких приложений является слишком узкоспециализированными[1].
1.1 WEB-приложения, используемые для анализа последовательностей ДНК
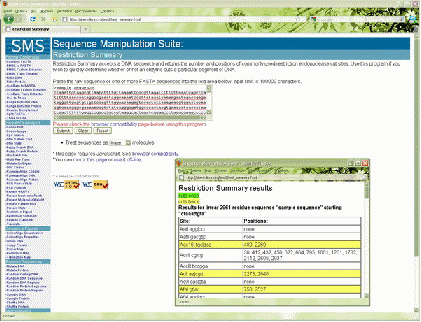
В качестве примера можно привести web-приложение Sequence Manipulation Suite [3]. Данное приложение работает on-line и включает в себя множество различных программ (более 50), которые значительно облегчают работу молекулярного биолога. Так, например, программа Restriction Summary (рисунок 1.1) позволяет быстро определить ферменты, разрезающие определенную последовательность ДНК; программа Reverse Translate преобразует аминокислотную последовательность белков в кодирующую данный белок нуклеотидную последовательность ДНК. Программа DNA Stats при вводе нуклеотидной последовательности ДНК и РНК определяет количество каждого азотистого основания в последовательности, а также процентное соотношение нуклеотидов и пар нуклеотидов, что может быть очень полезным для расчета температуры отжига праймеров.
Преимуществом данного web-приложения является его доступность и широкий выбор программ. Однако существует и ряд недостатков. Данное приложение не позволяет одновременно выполнять в одном окне несколько программ. Еще одним недостатком может быть то, что приложение Sequence Manipulation Suite доступно только на английском языке.
Рисунок 1.1 – программа Restriction Summary в web-приложении Sequence Manipulation Suite.
1.2 Приложения в составе Vector NTI Suite
Vector NTI Suite – это достаточно удобный набор программных инструментов, предназначенных главным образом для анализа последовательностей. Инструменты разработаны компанией InfoMax Inc. для специалистов в области молекулярной биологии и позволяют изучать, визуализировать, манипулировать и конструировать биологические молекулы, а также хранить и предоставлять доступ к информации об этих молекулах [4].
При помощи инструментов Vector NTI Suite можно делать следующее:
Легко ориентироваться и осуществлять поиск в базе данных молекул и ферментов;
Разрабатывать стратегию рекомбинации и соответствующие протоколы;
Симулировать гель-электрофорез, анализировать результаты эксперимента;
Анализировать и манипулировать ДНК, РНК и белковыми молекулами;
Создавать или импортировать молекулы, энзимы, олигонуклеотиды, добавлять описание и дополнительную информацию.
Vector NTI позволяет генерировать протоколы для клонирования и амплификации, разрабатывать пробы и праймеры, расcчитывать фрагменты рестрикции и симулировать гель-электрофорез. Приложения Vector NTI Suite обеспечивают молекулярный анализ, выравнивание и сборку последовательностей из фрагментов. К базовым приложениям Vector NTI Suite следует отнести следующие программы и инструменты:
Основная программа Vector NTI (Графическая оболочка для работы с базой данных, молекулами, гель-электрофорезом, обеспечивающая интеграцию на уровне данных с другими приложениями Vector NTI Suite);
3Dmol - приложение, позволяющее эффективно визуализировать и манипулировать 3х-мерными молекулярными структурами, описанными в стандарте Protein Data Bank (программа по функциям близка к Rasmol);
AlignX – программа для выравнивания последовательностей (множествен.), позволяющая работать как с протеинами, так и с нуклеиновыми кислотами. AlignX читает все стандартные текстовые форматы, например FASTA, GenBank, EMBL, SWISS-PROT, GenPept и ASCII- текст;
AlignX Blocks – программа для определения положения, анализа и редактирования блоков из найденных подобий среди множества белковых последовательностей;
BioPlot – набор инструментов для анализа последовательностей белков и нуклеиновых кислот, предоставляющих более пятидесяти различных предопределенных градаций для анализа белковых последовательностей (ProtScale-анализ), связанных с картой свойств и последовательностью аминокислот. BioPlot позволяет выполнять восемь различных видов анализа нуклеиновых кислот, среди которых анализ: температуры плавления, свободной энергии, энталпии, энтропии, содержания GC;
ContigExpress – программа для манипулирования и сборки множества небольших фрагментов, как текстовых последовательностей, так и хроматограмм с автоматического секвенатора, в длинные последовательности;
GCG Converter – инструмент в составе Vector NTI Suite, который преобразует последовательности в файл формата GCG, так чтобы их можно было импортировать в Suite;
PubMed/Entrez Search – это поисковый механизм для поиска (и извлечения) цитат и молекул из публичных баз данных, таких как PubMed, GenBank и Protein DataBank. Результаты поиска могут быть импортированы другими приложениями Vector NTI Suite [4].
В терминах Vector NTI, конструирование – это создание молекул из полностью предопределенных фрагментов, сочетание которых определяется пользователем программы. Конструирование молекулы ДНК выполняется по следующей схеме:
Выбор и описание фрагментов молекулы;
Создание списка фрагментов, описывающего целевую молекулу (Goal Molecule Definition List);
Вызов соответствующего инструмента и собственно конструирование молекулы.
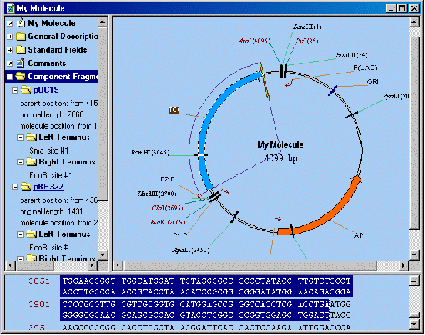
Молекула ДНК может быть собрана из различных типов фрагментов – из фрагментов существующих молекул, линкеров, адаптеров и т.п. (рисунок 1.2). Значительная часть работы при конструировании молекул - это определение фрагментов, описание линкеров и адаптеров занимает значительно меньше времени. К счастью, в составе Vector NTI есть специальный инструмент – Мастер фрагментов (Fragment Wizard), который "руководит" пользователем в процессе описания нового фрагмента и обеспечивает быстроту и прозрачность процедуры [5].
Рисунок 1.2 – Пример сконструированной плазмидной молекулы на основе векторов pUC19 и pBR322 в программе Vector NTI [5]
В дополнении к Fragment Wizard для определения фрагментов можно использовать Редактор фрагментов (Fragment Editor). С помощью редактора легко описываются любые фрагменты, однако наиболее эффективно он может быть использован при определении линкеров и адаптеров.
Кроме конструирования, в рамках инструментария Vector NTI для создания и добавления новой молекулы в базу данных существует целый ряд возможностей, в частности:
Описание молекулы может быть импортировано из GenBank, EMBL, FASTA;
Для описания молекулы может быть использована последовательность из какого-либо текстового файла;
Молекула может быть разработана по правилам встроенной в Vector NTI базы биологических знаний.
Молекулы, которые собраны из фрагментов, в терминах Vector NTI называются сконструированными молекулами. Молекулы, описание которых импортировано или составлено вручную, называются базовыми молекулами, т.к. они записываются в базу данных как нечто целое в противовес сконструированным молекулам, для которых вместе с общими данными записывается информация об исходных фрагментах [5].
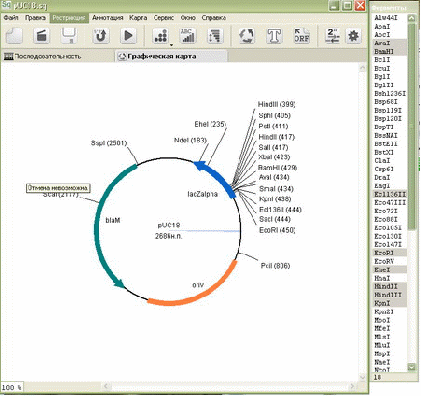
1.3 Компьютерная программа SQ для редактирования и анализа биологических последовательностей
Приложение SQ было разработано преподавателями кафедры молекулярной биологии биологического факультета БГУ, доцентом, к.б.н. Николайчиком Е. А. и к.б.н. Валентовичем Л.Н.
SQ – редактор последовательностей ДНК, предназначенный для создания, редактирования и анализа последовательностей ДНК, а также для моделирования экспериментов молекулярного клонирования и ПЦР. Простота и удобство интерфейса были одной из основных целей при разработке программы SQ. Для достижения этой цели в SQ реализованы поддержка альтернативных языков интерфейса, удобная панель инструментов и использование вкладок вместо отдельных окон для отображения результатов анализа последовательности (рисунок 1.3).
Рисунок 1.3 – Интерфейс компьютерной программы SQ
Программа SQ использует классификацию стандартных функциональных участков последовательностей ДНК, принятую в формате GenBank [6] и таблицы генетического кода [7], опубликованные на сайте NCBI.
При помощи программы SQ можно выполнить следующие операции:
Получить обратную, комплементарную или обратно-комплементарную цепь ДНК;
Поиск промоторных, операторных и прочих последовательностей;
Создание, отображение и редактирование аннотации последовательности ДНК;
Трансляция нуклеотидных последовательностей и поиск открытых рамок считывания;
Рестрикционный анализ;
Виртуальное клонирование;
Проверка праймеров для ПЦР.
Процедуры рестрикционного анализа используют базу данных REBASE [8]. Связанные с рестрикцией функции доступны через многие элементы интерфейса программы: через панель инструментов, стандартное меню, контекстные меню основного окна и вспомогательного окна "Ферменты". Последнее окно содержит список доступных ферментов, представляющий собой выборку из рестрикционных эндонуклеаз, описанных в базе данных REBASE [2]. Меню "Рестрикция" позволяет редактировать этот список, а также создавать/редактировать наборы ферментов. Также графическая карта предоставляет некоторые дополнительные возможности по сравнению с простым рестрикционным анализом[1].
Виртуальное клонирование позволяет моделировать реальные эксперименты по молекулярному клонированию. Последовательности рекомбинантных плазмид, которые можно создать с помощью этой методики, облегчают документацию экспериментов по молекулярному клонированию, а также помогают оценить размеры фрагментов рестрикции, ожидаемых при проверке рекомбинантных клонов[1].
Программа SQ является сегодня единственной, предназначенной для анализа нуклеотидных последовательностей и симуляции молекулярного клонирования программой с русскоязычными (а также белорусскоязычными) интерфейсом и документацией. Несмотря на то, что программа SQ уступает по своим функциональным возможностям аналогичным коммерческим продуктам, она превосходит большинство из них по удобству пользования (и, естественно, цене).Программа SQ распространяется свободно и не имеет ограничений по ее использованию. Последняя версия программы и вспомогательные файлы доступны для загрузки на официальном сайте проекта SQ [9].
Глава 2
Моделирование макромолекул и процессов протекающих на молекулярном уровне
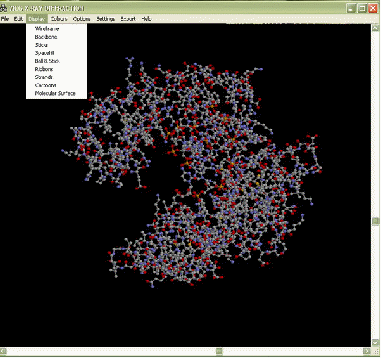
2.1 Программа RasWin для просмотра 3D молекул
Данная программа находится в свободном доступе в глобальной сети «Интернет». RasWin позволяет просматривать изображения молекул в формате .pdb. Файлы в данном формате можно найти в базе данных Protein Data Base (PDB) [10]. Для того чтобы найти интересующую структуру необходимо в окне поиска набрать код структуры или название, например, 132L - лизоцим. В открывшемся окне, найти ссылку на файл .pdb, который откроется в окне браузера. Далее необходимо скопировать весь текст в блокнот и сохранить как name.pdb (изменить при сохранении 'текстовые файлы' на 'все файлы', для того, чтобы изменить разрешение). Пользовательский интерфейс достаточно прост (рисунок 2.1).
Рисунок 2.1 – Интерфейс программы RasWin
При помощи RasWin макромолекулы можно представить в различных вариациях (рисунок 2.2). Также в данной программе предусмотрен выбор цветов для отдельных групп атомов, которые составляют макромолекулу. Полученные 3D изображения можно сохранить в форматах .bmp, .gif, .epsf, .ppm и .rast.
Рисунок 2.2 – различные варианты представления 3D модели молекулы ДНК полимеразы бета (код 7ICG в базе данных Protein Data Base) с помощью программы RasWin
2.2 Программа Modeller для моделирования макромолекул.
Modeller - компьютерная программа, которая моделирует трехмерные структуры белков и строит их со всеми необходимыми пространственными ограничениями. Modeller является наиболее часто используемой программой для гомологического или сравнительного моделирования структуры белков: пользователь предоставляет выровненную последовательность, на основе которой программой будет смоделирована молекула с известными родственными структурами. В процессе работы Modeller автоматически вычислит модель со всеми неводородными атомами.
В более широком смысле, использование программы Modeller ограничено пространственной структурой аминокислотной и лигандной последовательностью, которые будут смоделированы. Результат вычислений - трехмерная структура, которая соответствует этим ограничениям настолько, насколько это возможно [11].
Трехмерная модель, полученная в программе Modeller при оптимизации функции вероятности молекулярной плотности имеет формат (pdf). Молекулярная pdf для сравнительного моделирования оптимизирована с переменно-целевой процедурной функцией в пространстве декартовых координат, которое использует методы сопряженных градиентов и динамику молекул с модельной "закалкой". Программа Modeller может также выполнять множественное сравнение последовательностей белка и/или структур, объединенных в кластеры белков, и поиска последовательности из баз данных. Программа использует язык скриптов (сценариев) и не имеет графического интерфейса. Программа Modeller распространяется бесплатно. Скачать ее можно с официального сайта Modeller [12].
2.3 Использование программы AutoDock для моделирования молекулярных процессов
Докинг – одна из самых главных и важных стадий процесса компьютерного моделирования лекарств. Основной задачей докинга является построение модели структуры комплекса молекулы лиганда (биологически активного вещества) и молекулы рецептора (биомишени). Обычно молекула рецептора представляет собой белковую макромолекулу, а молекула лиганда - малую молекулу. Реже встречаются примеры белок-белкового докинга [13].
Молекулярный докинг предсказывает структуру межмолекулярного комплекса, сформированного между двумя или больше молекулами. При докинге необходимо учитывать конформационную подвижность как молекулы рецептора так и лиганда.
Молекулярный докинг рассматривается как размещение низкомолекулярного вещества в активный сайт белка и предсказание энергетически выгодного расположения. Когда активный сайт белка не известен, поиск сайт соединения и конформации лиганда называют "слепым докингом". Исследование при слепом докинге также ценны для поиска белок-белковых взаимодействий, когда обе макромолекулы, как предполагается, дополняют друг друга формой и межмолекулярными взаимодействиями. В слепом докинге предполагается, что энергетически выгодная конформация с учетом комплементарности является искомой структурой. Для решения данных задач применяется одна из наиболее эффективных программ – AutoDock [14] .
Программа AutoDock позволяет проводить молекулярный докинг с целью поиска локального минимума энергии взаимодействия между лигандом и белком, а также проводить поиск глобального минимума энергии взаимодействия между лигандом и белком, если положение лиганд связывающего центра не определено эксперементально. В частности, программа применяется для разработки лекарств, специфически связывающихся с тем или иным белком, при виртуальном высокопроизводительном скрининге, при белок-белковом докинге, а так же для изучения химических механизмов взаимодействия, для определения геометрии лиганд-рецепторных комплексов рецепторов на основе использования экспериментальных или расчетных данных о строении лиганд-связывающего центра. При расчете энергии связывания учитываются ван-дер-ваальсовы и электростатические взаимодействия, водородные связи, а также вклад энергии десольватации. Эффективность докинга в программе AutoDock повышается путем построения карт электростатических потенциалов, силового поля AMBER. Результаты, полученные с помощью программы Autodock, позволяют анализировать минимальную энергию связывания лиганда с макромолекулой, вероятность нахождения лиганда в активном центре белка в положении, отвечающем наименьшей энергии связывания, и среднеквадратичное отклонение найденного положения лиганда от данных рентгеноструктурного анализа [13,14].
AutoDock состоит из двух главных программ: AutoDock выполняющий докинг лигандов в сайт связывания мишений белка в решетке; AutoGrid предварительно вычисляет размер этой решетки. В дополнение к использованию существует GRID - распределение визуализированных моментов между этими двумя частями. Это может быть полезным, например, при дизайне синтетических органических соеденений, когда запросы программы очень массивные и идет большая нагрузка на процессор компьютера.
Также разработан графический пользовательский интерфейс, который называется AutoDockTools или сокращенно ADT, который позволяет визуализировать вращение лиганда, просматривать места взаимодействия лиганда и молекулеы управлять докингом.
Программа AutoDock применяется в:
Рентгеновской кристаллографии;
Дизайне структуры кандидатов на роль лекарств
Проведении оптимизации;
Виртуальном скрининге (HTS);
Дизайне комбинаторных библиотек;
Белок-белковых взаимодействий;
Механизмах химических исследований [13,14].
Современную науку трудно себе представить без информационных технологий. В данной работе были рассмотрены методы использования ИТ в молекулярной биологии. Постановка многих экспериментов в молекулярной биологии была бы крайне затруднительна без использования компьютерных устройств и программного обеспечения. Такие программы как Vector NTI, SQ позволяют изучать, визуализировать, манипулировать и конструировать биологические молекулы, разрабатывать стратегии клонирования молекул, тем самым в большой степени повышая продуктивность работы молекулярного биолога. Также при помощи целого ряда программ, таких RasWin, Modeller, AutoDock возможно не только 3D моделирование макромолекул, но процессов, идущих на молекулярном уровне.
Николайчик Е. А., Валентович Л. Н.. SQ –компьютерная программа для редактирования и анализа биологических последовательностей // Труды Белорусск. гос. ун-та. Сер.: Физиологические, биохимические и молекулярные основы функционирования биосистем. 010. –Т.5. –Ч.1. –с.57-67.
Rice, P., Longden, I., & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite // Trends in Genetics: TIG – 2000. – Vol. 16. – P. 276-277.
Sequence Manipulation Suite [Electronic resource] / University of Alberta. – Canada, 2004. – Mode of access: http://sherb.lin.irk.ru/sms2/index.html. – Date of access: 10.12.2010.
Хмелько, И. Приложения в составе Vector NTI Suite / Практическая молекулярная биология [Электронный ресурс].– 2007. – Режим доступа: http://molbiol.ru/protocol/09_09a.html. – Дата доступа: 10.12.2010.
Хмелько, И. Конструирование молекул / Практическая молекулярная биология [Электронный ресурс].– 2007. – Режим доступа: http://molbiol.edu.ru/review/03_02c.html. – Дата доступа: 15.12.2010.
Genetic Sequence Data Bank NCBI-GenBank Flat File Release 180.0 [Electronic resource] / National Center for Biotechnology Information – USA, 2010. – Mode of access: ftp://ftp.ncbi.nih.gov/genbank/gbrel.txt.– Date of access: 12.12.2010.
Elzanowski, A., Ostell, J. The Genetic Codes // National Center for Biotechnology Information [Electronic resource] – 2010. – Mode of access: http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi?mode=c. – Date of access: 12.12.2010.
Roberts, R. J., Vincze, T., Posfai, J., & Macelis, D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes // Nucleic Acids Research – 2010. – Vol. 38. – P. D234-236.
SQ: редактор биологических последовательностей для молекулярных биологов // Биологический факультет Белорусск. гос. ун-та [Электронный ресурс].– 2003. – Режим доступа: http://www.bio.bsu.by/sq/about.html. – Дата доступа: 10.12.2010.
A Resource for Studying Biological Macromolecules // Protein Data Bank [Electronic resource] – 2008. – Mode of access: http://www.rcsb.org/pdb/home/home.do. – Date of access: 19.12.2010.
Программа Modeller - что это и для чего? // Биоинформатика и познания [Электронный ресурс].– 2009. – Режим доступа: http://www.bioinformatix.ru/modeller/programma-modeller-chto-eto-i-dlya-chego.html. – Дата доступа: 19.12.2010.
Mollder [Electronic resource] / Departments of Biopharmaceutical Sciences and Pharmaceutical Chemistry, California Institute for Quantitative Biomedical Research, 2010. – Mode of access: http://salilab.org/modeller/download_installation.html. – Date of access: 19.12.2010.
Энгелевский Н. Докинг: определение, виды, методы, проблемы и решения. Программы для докинга / Биоинформатика и познания [Электронный ресурс].– 2009. – Режим доступа: http://www.bioinformatix.ru/drugie-programmyi/doking-opredelenie-vidyi-metodyi-problemyi-i-resheniya.-programmyi-dlya-dokinga.html. – Дата доступа: 19.12.2010.
Мартынюк А. Молекулярный докинг: понятие, применение, программы для докинга / Биоинформатика и познания [Электронный ресурс].– 2009. – Режим доступа: http://www.bioinformatix.ru/drugie-programmyi/molekulyarnyiy-doking-ponyatie-sut-programmyi-dlya-dokinga.html. – Дата доступа: 19.12.2010.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ К РЕФЕРАТУ
A
AutoDock, 14, 15, 16
G
GenBank, 8, 9, 11
M
Modeller, 13, 14, 16
R
RasWin, 12, 13, 16
S
SQ, 10, 11, 16
V
Vector NTI, 7, 8, 9, 10, 16
Б
база данных, 7, 8, 11, 12, 13, 14
белок, 4, 5, 6, 8, 13, 14, 15, 16
Д
ДНК, 4, 5, 6, 7, 8, 10, 11, 13
докинг, 14, 15
К
клонирование, 7, 10, 11, 16,
М
молекулярная биология, 4, 7, 10, 16,
П
праймер, 5, 7, 11
ПЦР, 5, 10, 11
Р
рестрикционный анализ, 5, 11,
Ф
фермент, 6, 7, 11,
ИНТЕРНЕТ РЕСУРСЫ В ПРЕДМЕТНОЙ ОБЛАСТИ ИССЛЕДОВАНИЯ
http://www.molbiol.edu.ru/
Практическая молекулярная биология. Сайт является незаменимым для биохимиков, генетиков, микробиологов и молекулярных биологов. Это крупнейшая биологическая база данных. Сайт содержит подробный справочник, который состоит из наиболее важных разделов. Здесь можно найти руководства и рекомендации по выполнению тех или иных операций, подробное описание методов исследования (работа с бактериями, бактериофагами, эукариотическими организмами, дигибридные системы, методы выделения и анализа ДНК про- и эукариотических организмов, работа с белками), методики и расчеты для приготовления растворов, подбор необходимых для исследования ферментов и реактивов. Можно следить за свежими публикациями. Имеются обзоры различных биологических ресурсов и программ, а также ссылки на биологические журналы и гранты биологического профиля. Внимание уделяется также образованию и образовательным ресурсам. Имеются сведения о компаниях и русскоязычных институтах биологического профиля, а также ссылки на полезные web-ресурсы. Также пользователи могут разместить на сайте используемые ими протоколы экспериментов, обзоры и дополнения к уже опубликованным материалам.
http://www.cellbiol.ru/
Информационный сайт-справочник по биологии и медицине. Он включает в себя образовательные материалы, которые могут быть полезны как школьникам, так и студентам биологических факультетов. Помимо образовательной информации, на сайте публикуются биографии известных ученых в области биологии, генетики, физиологии, медицины и многих других.
http://www.molbiol.ru/
Информационный проект, поддерживаемый русскоязычным биологическим сообществом. Здесь можно найти руководства и рекомендации по выполнению тех или иных операций, подробное описание методов исследования (работа с бактериями, бактериофагами, эукариотическими организмами, дигибридные системы, методы выделения и анализа ДНК про- и эукариотических организмов, работа с белками), методики и расчеты для приготовления растворов, подбор необходимых для исследования ферментов и реактивов. Можно следить за свежими публикациями. Имеются обзоры различных биологических ресурсов и программ, а также ссылки на биологические журналы и гранты биологического профиля. На этом сайте есть форум для его пользователей, в котором можно получить ответы на многие интересующие вопросы, касающиеся материалов представленных на данном сайте.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?DB=pubmed
PubMed — текстовая база данных медицинских публикаций на английском языке, на основе раздела биотехнология национальной медицинской библиотеки США (National Library of Medicine, NLM). База данных была разработана национальным центром биотехнологической информации (National Center for Biotechnology Information, NCBI). PubMed является бесплатной версией базы данных MEDLINE. PubMed документирует медицинские статьи из специальной литературы, а также дает ссылки на полнотекстовые статьи. PubMed включает в себя данные из разделов медицина, стоматология, ветеринария, общее здравоохранение, психология, биология, генетика, биохимия, цитология, биотехнология, биомедицина и т.д. Документированно около 3.800 биомедицинских изданий. Ежегодно база данных PubMed увеличивается на 500.000 документов. Поиск в базе данных журналов можно осуществлять по предмету или по названию журнала, по сокращенному названию, аббревиатуре ISO и другим параметрам.
http://searchlauncher.bcm.tmc.edu/
Search Launcher организован Baylor College of Medicine. Сайт предоставляет возможность проводить различные молекулярно-биологические анализы или поиски со стандартного и очень простого интерфейса. На сервере можно выполнять простые преобразования последовательностей. Есть возможность организовать анализ многих последовательностей одновременно. Ресурс просто незаменим для тех, кто не чувствует себя большим мастером по анализу последовательностей.
http://www.sciencekomm.at/
Более 4000 ссылок на биологические и медицинские журналы содержится на "science.komm" (там же удобные ссылки на полнотекстовые источники, словари, базы данных по абстрактам и т.п.). По web-ссылке вы попадаете на сайт конкретного журнала. На многих журналах можно подписаться на рассылку оглавления по E-mail.
http://www.sherb.lin.irk.ru/sms2/
Web-приложение Sequence Manipulation Suite, включающее в себя ряд полезных программ для работы молекулярного биолога. Сайт предоставляет возможность проводить различные молекулярно-биологические анализы.
http://www.bioinformatix.ru/
Сайт, посвященный биоинформатике, геномике, протеомике. На сайте публикуются статьи по этим тематикам. Кроме того, на данном сайте можно найти описание различных программ, используемых в биологии.
http://www.scirus.com/
Scirus – наиболее полная поисковая система для ученых в Интернете. Основанный на последних поисковых технологиях, он ищет более, чем в 300 миллионах определенных для науки Web-страницах, позволяя пользователям быстро находить: научные, медицинские и технические сведения; последние публикации; рецензируемые журналы; патенты и журналы, которые обычно пропускают другие поисковые системы. Эта поисковая система обращает внимание только на те Web-страницы, которые содержат научную информацию.
http://highwire.stanford.edu/
Этот сайт секции библиотеки Стэнфордского университета предлагает вниманию пользователей огромную базу материалов, которые можно загру-зить бесплатно в полном объеме. Источниками предлагаемых статей являются 975 журналов. Читатели имеют возможность доступа к полным текстам почти 1 435 924 статей, которые перед публикацией получили рецензию экспертов. Возможен быстрый поиск и расширенный поиск (по авторам статей, названи-ям, цитатам, ключевым словам и т.д.).
http://www.helgabad.narod.ru
Магистрантки Бадалян О.А. биологический факультет
Специальность «биология»
Смежные специальности
03.00.23 - биотехнология Генетические, селекционные и иммунологические исследования, изучение новых методов молекулярного клонирования генов для целей производства. Конструирование векторов, генов, рекомбинантных ДНК, гибридомная технология. Биотехнологии животных и растительных клеток; иммунная биотехнология. Генно-инженерные методы воспроизводства и селекции животных, получение трансгенных организмов.
03.00.15 - генетика Генетика биологических систем in vitro. Генетика клеточной дифференцировки. Экспрессия генов в культуре in vitro. Процессы андрогенеза и гиногенеза. Закономерности и механизмы изменчивости, индуцированной условиями in vitro. Создание нового генетического материала биотехнологическим и методами.
Основная специальность
03.00.26-молекулярная генетика
Экспрессия генов и ее регуляция.
Гены и регуляторные структуры, определяющие процессы транскрипции и трансляции.
Конструирование рекомбинантных молекул ДНК, синтез генов.
Молекулярные методы идентификации трансгенов, их экспрессия в генетически модифицированных организмах
Сопутствующие специальности
03.00.15 - микробиология
Выделение, культивирование и идентификация микроорганизмов.
ТЕСТОВЫЕ ВОПРОСЫ ПО ОСНОВАМ ИНФОРМАЦИОННЫХ ТЕХНОЛОГИЙ
ПРЕЗЕНТАЦИЯ МАГИСТЕРСКОЙ ДИССЕРТАЦИИ
11
22
33
44
55
66
77
88
99
110
111
СПИСОК ЛИТЕРАТУРЫ К ВЫПУСКНОЙ РАБОТЕ
С. Бондаренко, М. Бондаренко. Microsoft Word 2003 в теории и на практике.– Минск: Новое знание, 2004.–336с., ил.
М.В. Спека. Microsoft PowerPoint 2003: самоучитель.– Москва, Санкт-Петербург, Киев: Диалектика, 2004.–363 с., ил.
М.С. Шибут. Технологии работы с текстами и электронными таблицами: Word, Excel. Минск: общественное объединение «Молодежное научное общество», 2000.–142 с.

Нравится материал? Поддержи автора!
Ещё документы из категории разное:
Чтобы скачать документ, порекомендуйте, пожалуйста, его своим друзьям в любой соц. сети.
После чего кнопка «СКАЧАТЬ» станет доступной!
Кнопочки находятся чуть ниже. Спасибо!
Кнопки:
Скачать документ